
Vol 7 No 4 2022- 53

Computational discovery of novel anthelmintic natural compounds from Agave Brittoniana trel. Spp. Brachypus

Yeniel González-Castañeda,1 Yovani Marrero-Ponce,1-3* Jose O. Guerra,4 Yunaimy Echevarría-Díaz,1,2 Noel Pérez,5 Facundo Pérez-Giménez,3 Ana M. Simonet,6 Francisco A. Macías,6 Clara M. Nogueiras,7 Ervelio Olazabal,8 and Hector Serrano.8
Jose O. Guerra,4 Yunaimy Echevarría-Díaz,1,2 Noel Pérez,5 Facundo Pérez-Giménez,3 Ana M. Simonet,6 Francisco A. Macías,6 Clara M. Nogueiras,7 Ervelio Olazabal,8 and Hector Serrano.8
Jose O. Guerra,4 Yunaimy Echevarría-Díaz,1,2 Noel Pérez,5 Facundo Pérez-Giménez,3 Ana M. Simonet,6 Francisco A. Macías,6 Clara M. Nogueiras,7 Ervelio Olazabal,8 and Hector Serrano.81Universidad San Francisco de Quito, Grupo de Medicina Molecular y Traslacional (MeM&T), Escuela de Medicina, Colegio de Ciencias de la Salud (COCSA), Av. Interoceánica Km 12 1/2 y Av. Florencia, 17-1200-841 Quito, Ecuador.
2Departamento de Ciencias de la Computación, Centro de Investigación Científica y de Educación Superior de Ensenada (CICESE), Baja California 22860, Mexico.
3Unidad de Investigación de Diseño de Fármacos y Conectividad Molecular, Departamento de Química Física, Facultad de Farmacia, Universitat de València, Valencia, Spain.
4Chemistry Department, Faculty of Chemistry-Pharmacy. Universidad Central “Marta Abreu” de Las Villas, Santa Clara, 54830, Villa Clara, Cuba.
5Colegio de Ciencias e Ingenierías “El Politécnico”, Universidad San Francisco de Quito (USFQ), Quito, Ecuador.
6Grupo de Alelopatía, Departamento de Química Orgánica, Facultad de Ciencias, Universidad de Cádiz, C/República Saharaui, s/n, 11510 Puerto Real, Cádiz, España.
7Departamento de Química Orgánica, Facultad de Química, Universidad de La Habana, C/Zapata s/n entre G y Carlitos Aguirre, Vedado, Plaza de la Revolución, 10400, Ciudad de La Habana, Cuba.
8Chemical Bioactive Center. Universidad Central “Marta Abreu” de Las Villas, Santa Clara, 54830, Villa Clara, Cuba.
*Corresponding author. ymarrero@usfq.edu.ec or ymarrero77@yahoo.es; Tel.: +593-2-297-1700 (ext. 4021).
Available from: http://dx.doi.org/10.21931/RB/2022.07.04.53
ABSTRACT
Helminth infections are a medical problem in the world nowadays. This report used bond-based 2D quadratic indices, a bond-level QuBiLs-MAS molecular descriptor family, and Linear Discriminant Analysis (LDA) to obtain a quantitative linear model that discriminates between anthelmintic and non-anthelmintic drug-like organic-compounds. The model obtained correctly classified 87.46% and 81.82% of the training and external data sets, respectively. The developed model was used in a virtual screening to predict the biological activity of all chemicals (19) previously obtained and chemically characterized by some authors of this report from Agave brittoniana Trel. spp. Brachypus. The model identified several metabolites (12) as possible anthelmintics, and a group of 5 novel natural products was tested in an in vitro assay against Fasciola hepatica (100% effectivity at 500 µg/mL). Finally, the two best hits were evaluated in vivo in bald/c mice and the same helminth parasite using a 25 mg/kg dose. Compound 8 (Karatavinoside A) showed an efficacy of 92.2% in vivo. It is important to remark that this natural compound exhibits similar-to-superior activity as triclabendazole, the best human fasciolicide available in the market against Fasciola hepatica, resulting in a novel lead scaffold with anti-helminthic activity.
Keywords: TOMOCOMD-CARDD Software; QuBiLs-MAS, nonstochastic and stochastic bond-based quadratic indices; LDA-based QSAR model; Computational Screening, Anthelmintic Agent; Agave brittoniana Trel. spp. Brachypus, Fasciola hepatica.
INTRODUCTION
Helminths remain among the most common chronic infections, with more than one-third of the world’s population infected at any time.1 Currently, the high cost and toxicity of anthelmintics as well as the emergence of resistant strains of pathogenic helminths, have stimulated the desire to search for additional chemotherapeutic agents allowing a more efficient control of these parasites.2-4 A practical solution to this problem is to develop effective drugs from less expensive and more available raw materials.5 Natural products (NP) can be one of these materials for various reasons: 1) They inspired most of the active ingredients in medicines, 2) NP exhibit enormous structural diversity, 3) NP are the result of centuries of evolutionary pressure to create biologically active molecules, 4) the structural similarity of protein targets across many species, and so on. 5) It is extensively known that NP share more similar than synthetic compounds to the ‘chemical space’ of drug molecules.6-18 Unfortunately, only a small proportion of that diversity has been extensively explored for its pharmacological potential so far.19-21
Until now, the search for new anti-helminthic compounds from natural origin has generally been based on traditional trial-and-error methods.5,22 Unfortunately, these methods are highly inefficient and expensive.9,23 For this reason, new technologies have emerged to replace these old “hand-crafted” approaches for synthesis and testing new chemical entities.12,24-26 Virtual screening is an example of these modern approaches. Specifically, Quantitative Structure-Activity Relationships (QSAR) predictive models have been extensively used to filter large databases of compounds to identify new bioactive chemicals.27-34 Compared to other areas of pharmaceutical research; however, the screening of NPs has suffered from a lack of data in an appropriate format. Such information can significantly impact virtual screening, where new natural agents would be identified as potential therapeutic anthelmintics.
On the other hand, some authors of this report used an in-house computational approach to discover new anthelmintic synthetic compounds with rather good results35-37. A similar approach has been used to find new tyrosinase inhibitors from natural origin.38,39 However, no scientific report about discovering NPs with an anti-helminthic activity using an analogous computational strategy has been published.
This report presents the creation/validation of the QSAR model able to identify potential anthelmintic compounds. Next, we used this model in the virtual screening of NPs previously obtained and chemically characterized from Agave brittoniana Trel. spp. Brachypus. Finally, the identification/selection of the most promising anti-helminthic NPs for in vitro and in vivo experimental evaluation and the results of these evaluations are presented.
MATERIALS AND METHODS
Experimental Section
Materials. Compounds 1-5 were derived from previous studies made with Agave brittoniana Trel. spp. Brachypus.63 The rest of the chemicals were obtained using a similar approach described by the same research team.63 The extraction and purification of all compounds with a purity higher than 99% were carried out employing previously described methods.63 To obtain the initial dissolutions, each product dissolved in water at a concentration of 10 mg/mL (1%). The insoluble products were first dissolved in dimetylsulphoxide (DMSO) so that the concentrations of this product in the final solution did not exceed 1%. The necessary dilutions of each product to make possible the biological evaluation was obtained starting from the initial solutions. In addition, a solution of TCB was utilized as reference drug.
Animals. Healthy balb/c mice of both sexes (body weight: 0.018±0.001 Kg) and food were purchased from the National Center for Laboratory Animal Production (CENPALAB, Havana, Cuba). Quarantine, labeling, climatization and good maintenance conditions of animals were strictly obeyed.
General Experimental Procedures. To measure the chemical effectiveness against F. hepatica, an experimental technique reported in the literature was selected for biological material processing and F. hepatica egg extraction.66 Mitterpak et al.’s technique for the host (Lymnaea cubensis) invasion was carried out.67 Afterwards, we followed the steps reported by Olazábal et al.68 to obtain the metacercariae. Metacercariae were conserved in the cold until the in vivo experiment.66
Biological Experiments. The anthelmintic activity of the compounds was evaluated, first, against F. hepatica in an in vitro assay using an earlier described procedure and second, against metacercariae of the same pathogen in an in vivo experiment, applying another well-established procedure.
Several treatment groups with ten mice per group were created. One group (infected control group) was treated with Miglyol 810N (administration vehicle). The second group was neither infested nor treated. The remaining groups were treated with new chemicals. All mice received the new compounds through an oral route. Mouse invasion with metacercariae of F. hepatica, 2 weeks old, 14 days before drug administration, was carried out by Corba et al.’s method.69 The effectiveness was evaluated based on the following:
1) determination of the E% index. This is a quantitative indicator of effectiveness introduced by Steward70 and defined as E% = [(XC–XT)/XC] × 100.71 Here, E% is the percentage of effectiveness, XC is the average amount of Fasciola in the control group, and XT is the average amount of Fasciola in the treated group. Effectiveness was measured based on the elimination or not of F. hepatica, in its juvenile stage, as shown by laboratory diagnostics, using the helminthological necropsy on day 7 after the inoculated treatment.69
2) Determination of the hepatic index,72 by mean of the formula A = (B/C) × 100. In this case, A = hepatic index, B = liver weight and C = body weight.
3) Degrees of lesions of the liver.69
4) Spleen relative weight.73
5) Intensity of invasion making use of the formula I = A/B, where A = total amount of parasites, B = total amount of positives.
6) Extension of invasion by use of the formula %E.I = [T(t)/T(a)] × 100, where %E.I is the percent of invasion extensity, T(t) = number of total positives, and T(a) = total of infected animals.74
7) Gain of weight (final weight) (initial weight).
From these different effectiveness indexes,72-74 the E% index was selected.
Computational method
In the present report, we used a defined mathematical algorithm, which is characterized in this case by bond-based QuBiLs-MAS (acronym for Quadratic, Bilinear and N-Linear mapS based on graph–theoretic electronic-density Matrices and Atomic weightingS) MDs family (bond-level nonstochastic quadratic indices) to encode the chemical information in numbers.50-52 The CARDD extension of the TOMOCOMD approach has been previously successfully used to discover new bioactive molecular entities.35,36,38,44-49 The general principles of these indices and the main steps for the application of the QuBiLS-MAS50 software (http://tomocomd.com/software/qubils-mas) in QSAR/QSPR for drug design have been described in detail elsewhere.35,36,38,44-49
To find the classification function that discriminates between active and inactive compounds, we select the LDA because it is one of the most broadly used and straightforward techniques to obtain QSAR equations.35,36,48,49,75-85 It was carried out with the STATISTICA software.53 Forward-stepwise and best subset search procedures were fixed as the strategy for variable selection. The best model was selected considering the principle of parsimony (Occam’s razor). The considered tolerance parameter was the default value for minimum acceptable tolerance, which is 0.01. The quality of the model was determined by examining Wilks’ λ parameter (U statistic), the square Mahalanobis distance (D2), the Fisher ratio (F), and the corresponding p level [p(F)] as well as the percentage of good classification (accuracy) in the training and test sets (see Schemes 1 and 2). The classification of cases was performed by means of the posterior classification probabilities where one compound can then be classified as active if ΔP% > 0, being ΔP% = [P(Active) – P(Inactive)] >100, or as inactive otherwise. P(Active) and P(Inactive) are the probabilities with which the equation classifies a compound as active or inactive, respectively. On the other hand, the probability density approach implemented in the Ambit Disclosure software was used to evaluate the applicability domain of the model developed.60
RESULTS AND DISCUSSION
In silico study and virtual screening
Developing and validating linear QSAR models
To obtain a mathematical relationship between chemical structures and biological activity, the chemical information contained in many compounds must be statistically processed. Therefore, we build a data set containing 21240-43 and 30540,41 inactive compounds from the literature. It was build including 517 (active + inactive) compounds and was randomly divided into two subgroups: a set of 352 compounds (138 active and 214 inactive) that was used as the training set for developing the classification model and a second set of 165 compounds (74 active and 91 inactive) that was used as a test set for testing the predictive power of the model developed (see figure 1).
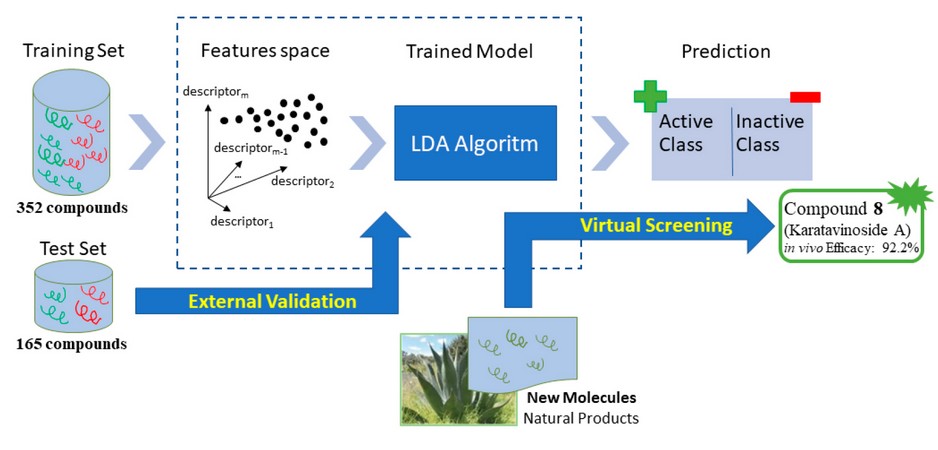
Figure 1. Schematic representation of the process used to design training and test sets.
Each structure was parameterized by using one TOMOCOMD-CARDD35,36,38,44-49 molecular descriptor (MDs) family, named bond-based nonstochastic 2D quadratic indices (QuBiLs-MAS Software)50-52 (see the experimental section for more details). Linear discriminant analysis (LDA), implemented on the STATISTICA software, was used as the statistical technique for model building.53 The best classification model obtained is given below, together with the LDA-statistical parameters:

where, N is the number of compounds, λ is the Wilks’ statistic, D2 is the squared Mahalanobis distance and F is the Fisher ratio.
The Wilks’ parameter is equal to the proportion of the total variance in the discriminant scores not explained by differences among the groups. Smaller values of Wilks’ lambda indicate the greater discriminatory ability of the function. Its statistic parameter can take values in the range of 0 (perfect discrimination) to 1 (no discrimination).54 That is, Wilks’ lambda is a direct measure of the proportion of variance in the combination of dependent variables unaccounted for by the independent variable (the grouping variable). Suppose a large proportion of the variance is accounted for by the independent variable. In that case, it suggests an effect from the grouping variable and that the groups (active and inactive) have different mean values. The Mahalanobis distance is a statistical technique that can be used to measure how distant a point is from the centre of a multivariate normal distribution, and its parameter indicates the separation between the respective groups.55 It shows whether the model has an appropriate discriminatory power for differentiating between the two respective groups. The classification of cases was carried out by means of the posterior classification probabilities. Using the Mahalanobis distances to do the classification, we can now derive probabilities. The probability that a case belongs to a particular class is basically proportional to the Mahalanobis distance from that group centroid. In summary, the posterior probability is the probability, based on our knowledge of the values of other variables, that the respective case belongs to a particular group.
This equation can correctly classify 87.46% (307/352) of the compounds in the training set and showed values of the Matthews correlation coefficients of 0.74 on it. More important, the model achieves a balanced classification accuracy in each group.
The results of the most relevant statistical parameters for this model are presented in Table 1, and the classification of compounds in the training set using Eq. 1 is presented in Table 2.

Table 1. Prediction Performances and Statistical Parameters for QSAR Models in the Training and Test Sets.
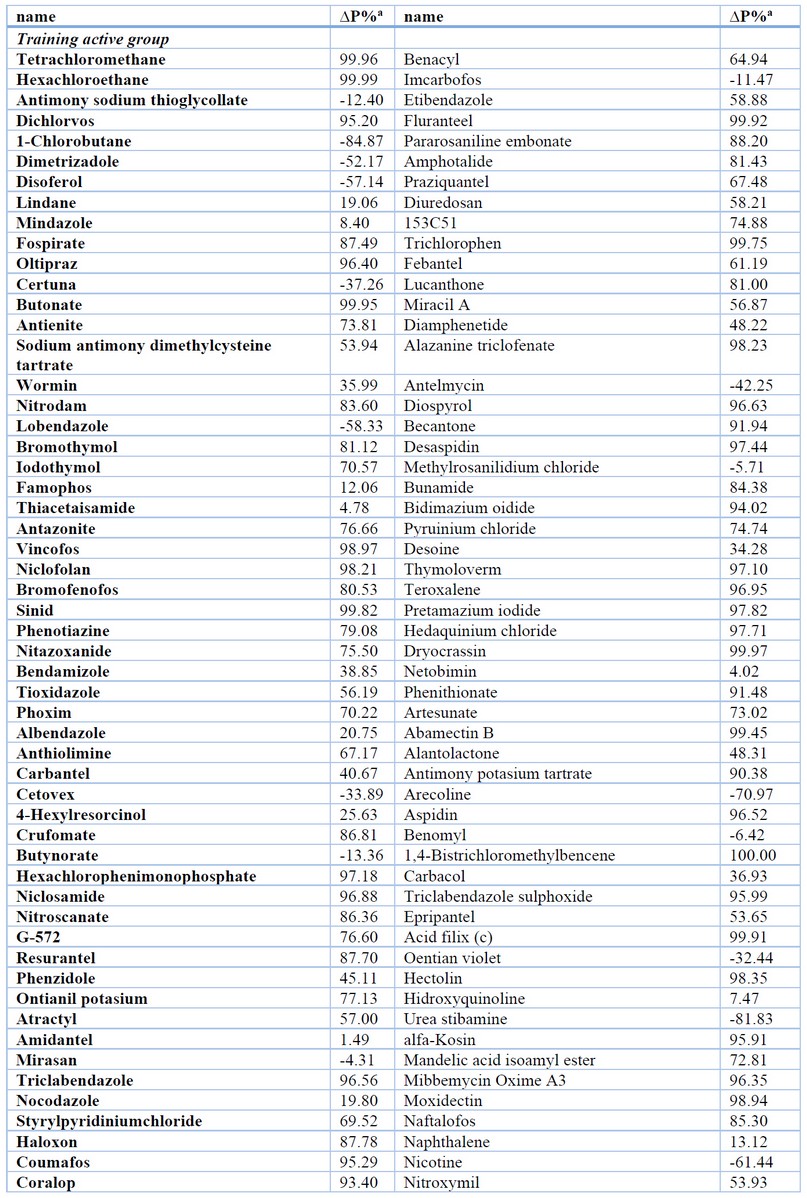




Table 2. Results of the Classification of Compounds in the Training and Test Set using QSAR Models.
Once a model is trained, its validation is another crucial aspect in this kind of analysis which can be performed by internal and external validation techniques (see Scheme 2).56,57 Here, a leave-many-out (LMO) cross-validation technique was carried out where groups of 176, 117, 70, 35, and 17 compounds of the training data (352 chemicals) were taken like cancellation groups and at each step. Then, the newly trained model was used to predict the left-out compounds. The results of this analysis are shown in Table 3, and the model’s parameters and predictions are rather stable when a perturbation is applied to the training set. This proofs that our model is robust.
In addition, to check the possibility of random correlations, the Y-randomization test (Y-scrambling) was performed by calculating the quality of the model randomly modifying the sequence of the response vector y (binary response: active or inactive) of the 5%, 10%, 20%, 30% y 40% of the compounds in the training set and recalculating the statistical parameters of the obtained models.57 The final conclusions of this test are present in Figure 2, indicating that the achieved level of random correlation is significantly lower than the original regression, leading to the conclusion that the models are not random.


Figure 2. Chemical Structures of Compounds Evaluating in the in silico Experiment from Agave brittoniana Trel. spp. Brachypus.

Table 3. Results of the Leave-Many-Out (LMO) Cross-Validation Analysis.
A more strict performance evaluation of a model is provided by an external validation where the model predictively is a challenge by compounds (external test set) that were not used in the model training (see Figure 3).57 Therefore, the equation obtained was evaluated in the test set (external prediction), showing accuracies of 81.82 % (135/165) and values of the Matthews correlation coefficients of 0.64. In addition to the external validation, the results of the statistical parameters described in Table 1 show that our model is not only robust but also predictive; therefore, it can be used in ligand-based virtual screening. The classification of both compounds in the external prediction set are depicted in Table 2.

Figure 3. General overview of the computational procedure.

Figure 4. Behavior of the Percentage of Good Classification in the Y-scrambling Analysis.
Finally, to define the applicability domain57 of Eq 1, a city-block distance-based approach58,59 implemented in the Ambit program60 was used. The model’s applicability domain was defined from the training set, and all compounds belonging to the external test series were inside it.
In silico identification of active compounds from natural products. Taking into consideration that NPs have inspired most of the active ingredients in medicines,10 in the last years a number of recent investigation was carried out to discover new active compounds from the natural origin using computational strategy.61 In our research, the developed model (Eq. 1) was used to filter an extensive database of NPs. All details of this database and other active (anthelmintics) NPs discovered by using our approach will be shown in the following reports.
Here, we only present the discovery of novel anthelmintic compounds from Agave brittoniana Trel. spp. Brachypus: a plant that grows like one of two endemic subspecies (ssp. Brachypus and ssp. Spirituana) of Agave brittoniana Trel. in the central region of Cuba.62 A group of nineteen compounds composed by 12 steroidal saponins, 6 steroidal sapogenins and 1 phytosterol (see Figure 4) that have been previously obtained and chemically characterized from this subspecies of Agave was evaluated in silico using the Eq. 1. These compounds were: agabrittonosides A–D,63 agabrittonosides E–K, karatavioside A,64 Diosgenin, Chlorogenin, Hecogenin, Tigogenin, Rockogenin, and β-Sitosterol.
As result of this virtual screening, twelve compounds were identified by the model as potential anti-helminthic hits (see Table 4).

Table 4. Results of the in silico Classification and Percentages of Anthelmintic Activity of the Selected Compounds from Agave brittoniana Trel spp. Brachypus in vitro and in vivo Assayed.
However, it is generally acknowledged that QSARs are valid only within the same domain for which they were developed. Even if the models are developed on the same chemicals, the DA for new chemicals can differ from model to model, depending on the specific MDs. One of the present reports aims is to develop a model for predicting the anthelmintic activity of NP at the early stages of the drug discovery and development pipelines. Therefore, the chemicals selected in this study were only evaluated in vitro after plotting them into the model’s previously obtained AD. In this analysis, all compounds were inside the DA of the model, ensuring excellent reliability for the prediction of this kind of lead used in the virtual screening. Moreover, all new leaders fall within the model’s DA, so the predictions are reliable.
Experimental corroboration
In vitro assay. Compounds were limited in availability; therefore, not all compounds were experimentally tested. Only three of the compounds detected in silico as potential anti-helminthic hits (Karataviosido A, Agabrittonósido A, Agabrittonósido B) and a mixture of Agabrittonósidos D and Agabrittonósidos E could be tested in vitro against F. hepatica at 5×10-1, 5×10-2, 5×10-3, 5×10-4, 5×10-5 and 5×10-6 mg/mL. Triclabendazole (TCB) was included in this experiment as a reference drug because it is the one of choice in treating human fascioliasis.65 Besides, Yucagenin (predicted as inactive) was also included in determining the influence of the glycoside moiety in the anti-helminthic activity. The biological in vitro evaluation results can also be seen in Table 4.
The experimental results agreed with the virtual screening predictions. As predicted, Yucagenin is not active at any test concentrations. However, its glycoside derivative (8, 9) had a bioactivity profile as TCB. This first saponin (8) has a glycoside rest joined to the C-3 atom identical to compound 9, its structural difference in the opening of the ring F and the glycosidation in the C-26 atom. The responsible for the little activity of 10, can be this structural modification or the increase of polarity of this zone. The mixture of compounds 12 and 13 presented in vitro activity higher than that observed for TCB. Compounds 12 and 13 are very similar structurally; both have the diosgenin-like central scaffold, but in compound 13 one xylose unit in 12 is substituted by a rhamnose group. In addition, 12 have a hydroxyl moiety in C-2, which is the only difference from 9. The combination of these subtle changes notably increases the activity of 12 and 13 concerning 9.
In vivo assay. An in vivo experiment using Bald/c mice-like biological models was conducted to obtain more profound conclusions about the pharmacological activity of in vitro hits. In this case, we only include in this experiment the two more active and pure substances (8 and 9) at doses of 3 mg/Kg. Table 4 shows the results of this study, where compound 8 was more active (92.16 % of efficacy) than 9 (52.94 %). The in vivo efficacy of compound 8 was identical to that of the control TCB. It is important to emphasize that this experiment was performed with a reduced dose (3 mg/kg). For instance, the TCB (the best human fasciolicide on the market65) is only wholly effective at 10 mg/kg. In addition, the few injuries in the livers and low inflammation of the spleens observed during the postmortem examination are qualitative criteria that positively appraise the effect of the tested compounds.
CONCLUSION
Today virtual screening has become an essential tool in drug discovery protocols. Here, bond-level quadratic indices (QuBiLs-MAS software, http://tomocomd.com/software/qubils-mas) and LDA were used to obtain a QSAR model that discriminates anthelmintic from inactive ones. Virtual screening of several metabolites from Agave brittoniana Trel. spp. Brachypus was carried out to discover new lead scaffold anthelmintics, and experimental corroboration showed that Karatavinoside A (8) exhibits similar-to-superior activity as triclabendazole (fasciolicide reference drug), with 100% in vitro effectivity (at 500 µg/mL) against Fasciola hepatica and 92.2% in vivo efficacy (25 mg/kg). This natural compound has been identified as a promising starting point for the rational optimization/design of new chemical derivatives with more potent anthelmintic activity.
Program availability: The QuBiLS-MAS software (portable standalone) and the respective user manual are freely available online at http://tomocomd.com/software/qubils-mas50
Acknowledgments. One of the present authors (M-P. Y) thanks the program ‘Estades Temporals per a Investigators Convidats’ for a fellowship to work at Valencia University (2020). Y.M.-P. and Noel Pérez acknowledge the support from Collaboration Grant 2019–2020 (Project ID16897) and Med Grant 2019-2020 (Project ID16911).
Competing interests: The authors declare no conflict of interest.
Author Contributions Statement: YG-C, FP-G, JOG, YE-D, NP and YM-P proposed the computational applications, QSAR modeling and performed the statistical analysis as well as prepared the manuscript. AMS, JOG, FAM, and CMN worked in the chemical methods and prepared the manuscript. EO and HS worked in the Parasitology tests. YG-C, FP-G, YE-D, NP and YMP worked in the QSAR modeling and performed the statistical analysis. All authors read and approved the final manuscript.
REFERENCES
1. Bundy DA, de Silva NR. Can we deworm this wormy world? . Br Med Bull. 1998;54:421–432.
2. Hammond JA, D. Fielding, S. C. Bishop. Prospects for plant anthelmintics in tropical veterinary medicine. Vet Res Comm. 1997;21:213-228.
3. WALLER PJ. Sustainable helminth control in developing countries. Vet Parasitol. 1997;71:195-207.
4. Monteiro AM, S. W. Wanyangu, D. P. Kariuki, R. Brain, F. Jackson, Q. R. Mckellar. Pharmaceutical quality of anthelmintics sold in Kenya. Vet Rec. 1997;142:396-398.
5. Suleiman MM, M. Mamman, Y. O. Aliu, J. O. Ajanusi. Anthelmintic activity of the crude methanol extract of Xylopia aethiopica against Nippostrongylus brasiliensis in rats. Vet arhiv 2005;75 (6):487-495.
6. T H, RM B, H M, F R. Statistical investigation into the structural complementarity of natural products and synthetic compounds. Angew Chem Int Ed. 1999;38:643-647.
7. Harvey AL. Natural products as a screening resource. Current Opinion in Chemical Biology. 2007;11:480–484.
8. Feher M SJ. Property distributions: differences between drugs, natural products, and molecules from combinatorial Chemistry. J Chem Inf Comput Sci 2003;43:218-227.
9. Ana G. Maldonado JPD, Michel Petitjean, Bo-Tao Fan. Molecular similarity and diversity in chemoinformatics: From theory to applications. Molecular Diversity 2006 10:39–79.
10. Sneader W, ed Drug prototypes and their exploitation. Chichester, UK: Wiley 1996.
11. Newman DJ CG, Snader KM. Natural products as sources of new drugs over the period 1981–2002. J Nat Prod 2003;66:1022-1037.
12. Ojima I. Modern molecular approaches to drug design and discovery. Acc Chem Res. 2008;41(1):2–3.
13. Clardy JW, C. Lessons from natural molecules. Nature 2004;432:829–837.
14. Jensen WFPR. Developing a new resource for drug discovery: marine actinomycete bacteria. Nature Chemical Biology. 2006 2 (12):666-673.
15. (WHO). World Health Organization Traditional Medicine Strategy 2002:2000-2005.
16. De Smet PA. The role of plant-derived drugs and herbal medicines in healthcare. Drugs. 1997;54:801-840.
17. Pezzuto JM. Plant-derived anticancer agents. Biochemical Pharmacology. 1997;53:121-133.
18. Cragg GM, Newman, D. J., Snader, K. M. Natural products in drug discovery and development. J Nat Prod. 1997 60 52-60.
19. Thomas M. Ehrman DJB, Peter J. Hylands. Phytochemical Databases of Chinese Herbal Constituents and Bioactive Plant Compounds with Known Target Specificities. J Chem Inf Model. 2007;47:254-263.
20. Harvey A. Strategies for discovering drugs from previously unexplored natural products. Drug Discovery Today 2000;5:294-300.
21. Tulp M BL. Unconventional natural sources for future drug discovery. Drug Discov Today 2004;9:450-457.
22. Adamczeski M, Quinoa E, Crews P. Unusual anthelminthic oxazoles from a marine sponge. Journal of the American Chemical Society. 1988;110(5):1598-1602.
23. Sadowski J KH. A scoring scheme for discriminating between drugs and non drugs. J Med Chem. 1998;41 3325–3329.
24. Jun Xu. AH. Chemoinformatics and Drug Discovery. Molecules. 2002;7:566-600.
25. Koehn FEC, G.T. The evolving role of natural products in drug discovery. Nat Rev Drug Discov. 2005; 4:206–220
26. Green MHR. Chemoinformatics – a new name for an old problem? Current Opinion in Chemical Biology 1999;3:379-383.
27. Zhang QYW, J.; Xu, X.; Yang, G.F.; Ren, Y.L.; Liu, J.J.; Wang, H.; Guo, Y. J Comb Chem. 2007;9:131-138.
28. Söderholm AAL, P.T.; Nyronen, T.H. J Med Chem. 2006;49:4261-4426.
29. Cherkasov A, Ban F, Li Y, Fallahi M, Hammond GL. J Med Chem. 2006;49:7466-7478.
30. Afantitis AM, G.; Sarimveis, H.; Koutentis, P.A.; Markopoulos, J.; Igglessi-Markopoulou, O. J Comput Aided Mol Des. 2006;20:83-95.
31. Lo Piparo EK, K.; Chana, A.; Benfenati, E. J Med Chem. 2006;49:5702-5709.
32. Shen MB, C.; Golbraikh, A.; Stables, J.P.; Kohn, H.; Tropsha, A. J Med Chem. 2004;47:2356-2364.
33. Alexander Hillebrecht GK. Use of 3D QSAR Models for Database Screening: A Feasibility Study J Chem Inf Model. 2008;48(2):384 -396.
34. S. Vadivelan BNS, Sheeba Jem Irudayam, Sarma A. R. P. Jagarlapudi Virtual Screening Studies to Design Potent CDK2-Cyclin A Inhibitors J Chem Inf Model. 2007;47(4 ):1526 -1535.
35. Marrero-Ponce Y, Castillo-Garit JA, Olazabal E, et al. TOMOCOMD-CARDD, a novel approach for computer-aided ‘rational’drug design: I. Theoretical and experimental assessment of a promising method for computational screening and in silico design of new anthelmintic compounds. Journal of Computer-Aided Molecular Design. 2004;18(10):615-634.
36. Marrero-Ponce YC-G, J. A.; Olazabal, E.; Serrano, H. S.; Morales, A.; Castañedo, N.; Ibarra-Velarde, F.; Huesca-Guillen, A.; Jorge, E.; Sánchez, A. M.; Torrens, F.; Castro, E. A. Atom, Atom-Type and Total Molecular Linear Indices as a Promising Approach for Bioorganic & Medicinal Chemistry: Theoretical and Experimental Assessment of a Novel Method for Virtual Screening and Rational Design of New Lead Anthelmintic. Bioorg Med Chem 2005;13(4):1005-1020.
37. Ibarra-Velarde F, Vera-Montenegro Y, Huesca-Guillen A, Canto-Alarcon Y, Alcala-Canto Y, Marrero-Ponce Y. In Silico Fasciolicide Activity of Three Experimental Compounds in Sheep. Animal Biodiversity and Emerging Diseases: Ann NY Acad Sci. 2008;1149:183-185.
38. Marrero-Ponce YK, M. T. H.; Casañola-Martin, G. M.; Ather, A.; Sultankhodzhaev, M. N.; Torrens, F.; Rotondo, R. Prediction of Tyrosinase Inhibition Spectra for Chemicals Using Novel Atom-Based Bilinear Indices. ChemMedChem. 2007;2(4):449-478.
39. Marrero-Ponce Y, Khan MT, Martín GM, et al. Atom-Based 2D Quadratic Indices in Drug Discovery of Novel Tyrosinase Inhibitors: Results of In Silico Studies Supported by Experimental Results. QSAR Comb Sci. 2007;26(4):469.
40. Negwer M. Organic-chemical drugs and their synonyms. In: Berlin A, ed1987.
41. The Merck Index. In 12th ed1996. Hall Ca, ed.
42. Martin RJ, Robertson, A. P, Bjorn, H. Parasitology 1997:114-111.
43. Martin R. Modes of action of anthelmintic drugs. Review. The vet j. 1997.
44. Vega MC, Montero-Torres A, Marrero-Ponce Y, et al. New ligand-based approach for the discovery of antitrypanosomal compounds. Bioorganic & Medicinal Chemistry Letters. 2006;16(7):1898-1904.
45. Marrero-Ponce Y, Machado-Tugores Y, Pereira DM, et al. A computer-based approach to the rational discovery of new trichomonacidal drugs by atom-type linear indices. Current Drug Discovery Technologies. 2005;2(4):245-265.
46. Casañola-Martín GM, Marrero-Ponce Y, Khan MTH, et al. TOMOCOMD-CARDD descriptors-based virtual screening of tyrosinase inhibitors: Evaluation of different classification model combinations using bond-based linear indices. Bioorganic & Medicinal Chemistry. 2007;15(3):1483-1503.
47. Marrero-Ponce Y, Iyarreta-Veitía M, Montero-Torres A, et al. Ligand-based virtual screening and in silico design of new antimalarial compounds using nonstochastic and stochastic total and atom-type quadratic maps. J Chem Inf Model. 2005;45(4):1082-1100.
48. Marrero-Ponce YM-M, R.; Torrens, F.; Martinez, Y.; Romero-Zaldivar, V.; Castro, E. A. . Atom, Atom-type, and Total Nonstochastic and Stochastic Quadratic Fingerprints: A Promising Approach for Modeling of Antibacterial Activity. Bioorg Med Chem. 2005;13:2881-2899.
49. Marrero-Ponce Y, Marrero RM, Torrens F, et al. Nonstochastic and stochastic linear indices of the molecular pseudograph’s atom-adjacency matrix: a novel approach for computational in silico screening and “rational” selection of new lead antibacterial agents. Journal of Molecular Modeling. 2006;12(3):255-271.
50. Valdés-Martiní JR, Marrero-Ponce Y, García-Jacas CR, et al. QuBiLS-MAS, open source multi-platform software for atom- and bond-based topological (2D) and chiral (2.5D) algebraic molecular descriptors computations. Journal of Cheminformatics. 2017;9(1):35.
51. Marrero-Ponce Y, Torrens F, Alvarado YJ, Rotondo R. Bond-based global and local (bond, group and bond-type) quadratic indices and their applications to computer-aided molecular design. 1. QSPR studies of diverse sets of organic chemicals. Journal of Computer-Aided Molecular Design. 2006;20(10):685-701.
52. Castillo-Garit JA, Marrero-Ponce Y, Torrens F, Garcia-Domenechc R, Rodriguez-Borges JE. Applications of Bond-Based 3D-Chiral Quadratic Indices in QSAR Studies Related to Central Chirality Codification. QSAR Comb Sci. 2009;28:1465 – 1477.
53. STATISTICA (data analysis software system) [computer program]. Version ver. 6.0. Tulsa: StatSoft, Inc; 2001.
54. Marrero-Ponce Y, Teran JE, Contreras-Torres E, et al. LEGO-based generalized set of two linear algebraic 3D bio-macro-molecular descriptors: Theory and validation by QSARs. Journal of Theoretical Biology. 2020;485:110039.
55. Etherington TR. Mahalanobis distances and ecological niche modelling: correcting a chi-squared probability error. (2167-8359 (Print)).
56. Paola Gramatica PP. Evaluation of Different Statistical Approaches for the Validation of Quantitative Structure-Activity Relationships2004, Italy.
57. OECD. Guidance document on the validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] models. In. Vol No. 69. Paris: OECD Environment Health and Safety Publications; 2007.
58. Jaworska J, Aldenberg T, Nikolova N. Review of methods for assessing the applicability domains of SARs and QSARs. Final report to the Joint Research Centre (Contract No. ECVA-CCR. 496575-Z). Part 1: Review of statistical methods for QSAR AD estimation by the training set. 2005; http://ecb.jrc.it/QSAR/Documents Accessed 2009/1/21, 2009.
59. Netzeva T, Worth AP, Aldenberg TA, et al. Current Status of Methods for Defining the Applicability Domain of (Quantitative) Structure–Activity Relationships. The Report and Recommendations of ECVAM Workshop 52. 2005.
60. AmbitDiscovery-v0.04 [computer program]. 2006.
61. Ronald J. Quinn ARC, Ngoc B. Pham, Paul Baron, Meredith E. Palframan, Lekha Suraweera, Gregory K. Pierens, Sorel Muresan. Developing a Drug-like Natural Product Library. J Nat Prod. 2008;71 (3):464–468.
62. Zayas Ad. Bol Soc Bot México. 1995;7:37.
63. Macías FA, Guerra JO, Simonet AM, Nogueiras CM. Characterization of the fraction components using 1D TOCSY and 1D ROESY experiments. Four new spirostane saponins from Agave brittoniana Trel. spp. Brachypus. Magn Reson Chem. 2007;45:615–620.
64. Vollermer YS GM, Gorovits TT, Abubakirov NK. Khim Prir Soedin. 1978; 6: 740.
65. Brunton LL, ed Goodman & Gilman’s the Pharmacological basis of Therapeutics 11th ed. California The McGraw-Hill Companies; 2006. John S Lazo KLP, ed.
66. Milligen FJ, Cornelissen, J.B., Guasenbeek, C.P. ,Bokhout, B.A.,. J Immunol Met. 1998 213:183.
67. Mitterpak J, Mendéz, M. and Mauri, M. Serie biológica. 1972 30 1.
68. Olazábal E, Morales, A., Serrano, H. and Brito, E. Vet Méx. 1999 30 109.
69. Corba J, Velebny S, Spaldonova R. Helminthology. 1981;18:43.
70. Steward JS. Parasitology. 1955;45:231.
71. Wood IB, Amaral NK, Bairden K, et al. Vet Parasitol. 1995;58:181.
72. Tsocheva N, Krustev L, Polyakova O. Helminthology. 1992;27:261.
73. Poitou I, Baeza E, Boulard C. Vet Parasitol. 1992;45:59.
74. Espaine L, Lines R, Demedio J. Manual de Parasitologıa y enfermedades Parasitarias I. In. Habana, Cuba: Editorial MES 1996:103.
75. González-Díaz HM-P, Y.; Hernández, I.; Bastida, I.; Tenorio, E.; Nasco, O.; Uriarte, U.; Castañedo, N.; Cabrera, M. A.; Aguila, E.; Marrero, O.; Morales, A.; Pérez, M. . 3D-MEDNEs: An Alternative “In Silico” Technique for Chemical Research in Toxicology.1. Prediction of Chemically Induced Agranulocytosis. Chem ResToxicol. 2003;16(10):1318-1327.
76. Julián-Ortiz JVdA, C. G.; Ríos-Santamarina, I.; García-Doménech, R.; Gálvez, J. Prediction of Properties of Chiral Compounds by Molecular Topology. J Mol Graphics Modell. 1998;16:14-18.
77. Marrero-Ponce Y. Total and local (Atom and Atom-Type) molecular quadratic indices: Significance-interpretation, comparison to other molecular descriptors and QSPR/QSAR applications. Bioorg MedChem. 2004;12:6351-6369.
78. Marrero-Ponce YC-G, J. A.; Torrens, F.; Romero-Zaldivar,V.; Castro, E. . Atom, atom-type and total linear indices of the “molecular pseudograph’s atom adjacency matrix”: Application to QSPR/QSAR studies of organic compounds. Molecules. 2004;9:1100-1123.
79. Marrero-Ponce YC, M. A.; Romero, V.; González, D. H.;Torrens, F. . A new topological descriptors based model for predicting intestinal epithelial transport of drugs in Caco-2 cell culture. J Pharm Pharm Sci 2004;7:186-199.
80. Marrero-Ponce YC, M. A.; Romero-Zaldivar, V.; Bermejo, M.; Siverio, D.; Torrens, F. . Prediction of intestinal epithelial transport of drug in (Caco-2) cell culture from molecular structure using ‘in silico’ approaches during early drug discovery. Internet Electronics J Mol Des. 2005;4:124-150.
81. Marrero-Ponce YG-D, H.; Romero-Zaldivar, V.; Torrens,F.; Castro, E. A. . 3D-chiral quadratic indices of the “molecular pseudograph’s atom adjacency matrix” and their application to central chirality codification: Classification of ACE inhibitors and prediction of σ-receptor antagonist activities. Bioorg Med Chem. 2004;12:5331-5342.
82. Marrero-Ponce YH-G, A.; Ibarra-Velarde, F. . Quadratic Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix” and Their Stochastic Forms: A Novel Approach for Virtual Screening and in silico Discovery of New Lead Paramphistomicide Drugs-like Compounds. Journal of Molecular Structure: THEOCHEM. 2005;717:67-79.
83. Marrero-Ponce YM-T, A.; Romero-Zaldivar, C.; Iyarreta-Veitía, I.; Mayón Peréz, M.; García Sánchez, R. Non-stochastic and stochastic linear indices of the “molecular pseudograph’s atom adjacency matrix”: Application to “in silico” studies for the rational discovery of new antimalarial compounds. Bioorg Med Chem. 2005;13:1293-1304.
84. Marrero-Ponce YN, D.; González-Díaz, H.; Ramos de Armas, R.; Romero-Zaldivar, V.; Torrens, F.; Castro, E. . Nucleic acid quadratic indices of the “macromolecular graph’s nucleotides adjacency matrix”. Modeling of footprints after the interaction of paromomycin with the HIV-1 ψ-RNA packaging region. Int J Mol Sci. 2004;5:276-293.
85. Estrada EU, E.; Montero, A.; Teijeira, M.; Santana, L.; De Clercq, E. A novel approach for virtual screening and rational design of anticancer compounds. J Med Chem. 2000;43:1975-1985.
Received: January 25, 2022 / Accepted: October 22, 2022 / Published:15 November 2022
Citation: González-Castañeda Y, Marrero-PonceY, Guerra J O, Echevarría-Díaz Y, Pérez N, Pérez-Giménez F, Simonet A M, Macías F A, Nogueiras C M, Olazabal E, Serrano H. Computational discovery of novel anthelmintic natural compounds from Agave Brittoniana trel. Spp. Brachypus. Revis Bionatura 2022;7(4) 53. http://dx.doi.org/10.21931/RB/2022.07.04.53
Vol 9 No 4 2024

INDEXADA EN
INDEXADA EN
