
Vol 8 No 4 2023 – 25
Asociación entre enfermedad periodontal y enfermedad de Alzheimer
Association between periodontal disease and Alzheimer’s disease
Association between periodontal disease and Alzheimer’s disease

Andrea Tamara García-Vásquez 1, Sandy Ruth Vidal-Chávez 2, Miriam Anccasi-Zevallos 3, Gina Adaliz Franco-Quispe 4, Donald Ramos-Perfecto 5 y Manuel Antonio Mattos-Vela 6*
1 Facultad de Odontología, Universidad Nacional Mayor de San Marcos, Lima Perú; andrea.garcia@unmsm.edu.pe .
2 Facultad de Odontología, Universidad Nacional Mayor de San Marcos, Lima Perú; sandy.vidal@unmsm.edu.pe.
3 Facultad de Odontología, Universidad Nacional Mayor de San Marcos, Lima Perú; miriam.anccasi@unmsm.edu.pe .
4 Facultad de Odontología, Universidad Nacional Mayor de San Marcos, Lima Perú; gina.franco@unmsm.edu.pe .
5 Facultad de Odontología, Universidad Nacional Mayor de San Marcos, Lima Perú; dramosp@unmsm.edu.pe .
6 Facultad de Odontología, Universidad Nacional Mayor de San Marcos, Lima Perú; mmattosv@unmsm.edu.pe .
* Correspondence: mmattosv@unmsm.edu.pe; Tel.: +51 51990770787.
Available from: http://dx.doi.org/10.21931/RB/2023.08.04.26
RESUMEN
La periodontitis es una enfermedad crónica que genera un deterioro progresivo de la salud periodontal y se caracteriza por inflamación de la encía, sangrado, bolsa periodontal, movilidad dental, así como la presencia de bacterias periodontopatógenas, como la Porphyromonas gingivalis, Aggregatibacter actinomycetemcomitans, Fusobacterium nucleatum, entre otras, agravan el estado cognitivo de estos pacientes y según varios estudios apuntarían a una potencial relación con la enfermedad de Alzheimer; enfermedad neurodegenerativa que se identifica por ocasionar demencia, pérdida de la memoria y disfunción cognitiva; asociada a múltiples factores de riesgo. Se confirmó la relación entre la enfermedad periodontal y enfermedad de Alzheimer, donde la presencia de bacterias periodontopatógenas agrava el estado cognitivo de los pacientes con este tipo de demencia. Conclusión: existe una relación importante entre la enfermedad periodontal y el Alzheimer, sustentado en estudios observacionales.
Palabras clave: Enfermedades periodontales, Enfermedad de Alzheimer, Inflamación, Demencia.
ABSTRACT
Periodontitis is a chronic disease that generates a progressive’s periodontal health deteriore and characterized by gum inflammation, bleeding, periodontal pocket, dental mobility, as well as the presence of periodontal pathogenic bacteria, such as Porphyromonas gingivalis, Aggregatibacter actinomycetemcomitans, Fusobacterium nucleatum, among others, aggravates the cognitive status of the patients. According to several studies, it would point to a potential relationship between Alzheimer’s disease, a neurodegenerative disease identified as causing dementia, memory loss, and cognitive dysfunction, associated with multiple risk factors. Review of the association between periodontal disease and Alzheimer’s. important literature on the subject was used, which complements the theoretical framework of the review. the relationship was confirmed between periodontal disease and Alzheimer’s disease, where periodontal pathogenic bacteria’s presence aggravates the cognitive status of the patients with this type of dementia. Conclusion: exists an important relationship between periodontal disease and Alzheimer, based on previous research.
Keywords: Periodontal diseases, Alzheimer’s disease; Inflammation, Dementia.
INTRODUCCIÓN
En la población de la tercera edad, se puede identificar una mayor presencia de demencia y enfermedad de Alzheimer (EA). Se estima que más de 113 millones de personas a nivel mundial tendrán EA para el año 2050 1,2. Además, hay muy pocos estudios que evalúen las implicaciones de la EA en la salud bucal. Sin embargo, los estudios han determinado que la EA conduce a la pérdida del autocuidado; y en las últimas etapas, puede llevar a la pérdida de función motora, como dificultades en la higiene oral, lo que podría generar la enfermedad periodontal (EP)3.
La evidencia actual indica que las afecciones crónicas inflamatorias aumentan el riesgo de EA lo que indicaría si otras afecciones inflamatorias, como la periodontitis, pueden contribuir a la prevalencia e incidencia de la EA4,5.
Por lo tanto, debido a la importancia de este tema es necesario investigar la posible relación bidireccional entre EP y EA.
La EA es una patología neurodegenerativa altamente prevalente, incidente, con elevados índices de mortalidad y morbilidad. Desracando su gran impacto en la salud pública debido a los costos de atención, el impacto general en los cuidadores y la sociedad. La EP, después de la caries dental, es la segunda más prevalente en la cavidad bucal. Estudios han asociado a la microbiota oral de la EP con las alteraciones sistémicas que aumentan el riesgo de EA. Se sabe que, la microbiota oral libera mediadores inflamatorios que pueden causar neuroinflamación, cuando está presente de forma arraigada tiene la capacidad de afectar células y su prevención basada en el cuidado de la salud bucal es esencial para prevenir la disbiosis permanente. El objetivo de esta revisión es actualizar los conocimientos sobre la relación entre la EP y la progresión de la EA6-8.
Enfermedad periodontal
Disbiosis oral
La comunidad microbiana conocida como “biofilm” es considerada un proceso dinámico que empieza al nacer y aumenta considerablemente según la edad, las enfermedades de la boca se tornan más comunes mientras estas alcanzan su estado de maduración. En el biofilm existen dos tipos de colonizadores, los primarios o “pioneros” caracterizados por un predominio de bacterias grampositivas y los secundarios caracterizados por el predominio de bacterias gramnegativas encargados de formar una red de matriz de EPS (Matriz de Exopolisacárido) para que las bacterias presentes puedan reforzar su estructura antes de migrar a otras zonas ante la escasez de nutrientes. Si bien en la mayor parte de la cavidad oral predomina el Streptococcus, la microbiota oral varía según los diferentes ecosistemas primarios de la cavidad bucal. En pacientes con EP la prevalencia y el número de especies anaerobias aumentan. Se sabe que la cavidad oral es famosa por la gran variedad de ecosistemas que posee con medios tanto aerobios como anaerobios en estructuras como labios, lengua, carrillo, amígdalas, paladar, entre ellas los surcos subgingivales9, los cuales dependiendo de los factores como temperatura, pH, humedad y restos alimenticios brindan las condiciones adecuadas para que la microbiota oral presente pueda coexistir. A su vez, estas pueden estar influenciadas por factores físicos y ambientales que alteran el equilibrio, teniendo en cuenta que la coagregación y la cooperación metabólica son claves para su supervivencia en cavidad oral10. Socransky elaboró un esquema que clasifica a las bacterias en complejos bacterianos, y que, según su grado de patogenicidad, debido a poseer suficientes factores de virulencia, pueden llevar a generar la disbiosis el surco gingival, entre estos el complejo rojo es conocido por la presencia de bacterias periodontopatógenas, causantes de la EP11. El aumento de las enfermedades en boca como caries y EP se debe al desequilibrio de la microbiota oral, provocado por un aumento en el número de especies microbianas patógenas y para controlarlo es necesario mantener la integridad de la microbiota oral residente. Si bien el sistema inmune cumple un rol de regulación ante el desborde de la microbiota oral, jugando un papel importante en el mantenimiento de la salud bucal9,10.
Bacterias periodontopatógenas (BPP) e inflamación
La periodontitis es una enfermedad inflamatoria crónica multifacotiral, siendo uno de los factores que más destaca, la presencia de bacterias Gram negativas que habitan la zona subgingival del periodonto (Figura 1). Entre las descritas dentro del complejo rojo de la pirámide de Socransky se encuentran Porphyromonas gingivalis, Aggregatibacter actinomycetemcomitans, Tannerella forsythia, Treponema denticola, Prevotella intermedia Fusobacterium nucleatum11,12. La adición constante de especies durante la sucesión microbiana conducirá a un estado de inflamación gingival. A su vez, el aumento de la inflamación daría lugar a un mayor crecimiento de especies colonizadoras11, que al saturarse en el espacio subgingival atraviesan la BHE (barrera hematoencefálica), la cual se vuelve más permeable a medida que las personas envejecen y pasan al torrente sanguíneo, donde las BPP son capaces de liberar citoquinas de inflamación como IL-1α, IL-1β, IL-6, (factor de necrosis tumoración α (TNF-α), metaloproteínas (MMP) en el torrente sanguíneo. Estás interleuquinas (IL), son causantes de fenómenos inflamatorios locales y sistémicos y han sido relacionadas con la progresión de enfermedades crónicas como diabetes, cáncer gástrico, periodontitis, enfermedades coronarias, EA, entre otras12,13. Los estudios coinciden al sugerir que un estado de inflamación crónica podría conducir a una neuroinflamación que afecta a las células nerviosas, deteriorándolas, resultando en una enfermedad neurodegenerativa como es la EA12,13.
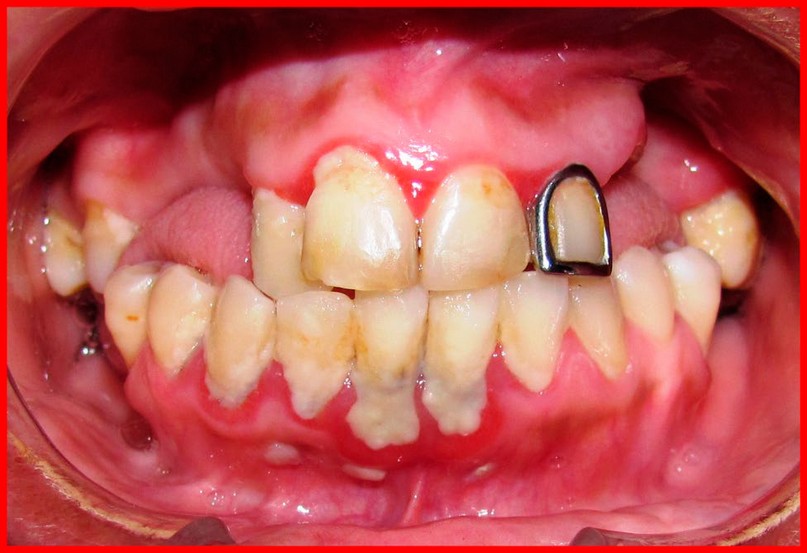
Figura 1. La enfermedad periodontal es un proceso infeccioso asociado principalmente con la presencia del biofilm dental, donde las estructuras afectadas son la encía, ligamento periodontal, hueso alveolar y cemento radicular.
Enfermedad de Alzheimer
La EA es la principal causa de demencia a nivel mundial, comprende entre un 60 y 70% de todos los casos14. Es un tipo de demencia degenerativa irreversible, que afecta a la memoria, cognición, comportamiento, capacidad de una persona para valerse por sí misma y realizar actividades cotidianas15. Las consecuencias del deterioro cognitivo y conductual a largo plazo podría incluso conducir a la muerte15.
Según datos de la Organización Mundial de la Salud (OMS), a nivel mundial 50 millones de personas viven con demencia, una proyección para el 2030 indica que habría 82 millones de casos, y para el 2050 esta cifra se duplicará, lo que indicaría que los casos aumentarán constantemente16. En EE. UU entre el 2000 y 2019 las muertes por EA aumentaron en un 145%17.
Las primeras manifestaciones sintomatológicas de la EA comprenden cambios conductuales inconscientes, pérdida de la memoria respecto a información nueva, deterioro del lenguaje y del habla. Las manifestaciones en estadios avanzados incluyen problemas de memoria episódica anterógrada, alucinaciones, desorientación y pérdida de la autonomía de la persona afectada15.
La EA se caracteriza, por presentar degeneración neuronal selectiva, disminución de los procesos sinápticos y presencia de lesiones a nivel cerebral específicamente, placas seniles o placas neuronales de β-amiloide (AβP) a nivel extracelular y ovillos neurofibrilares formados por proteínas Tau hiperfosforiladas a nivel intracelular15,18,19 (Figura 2).
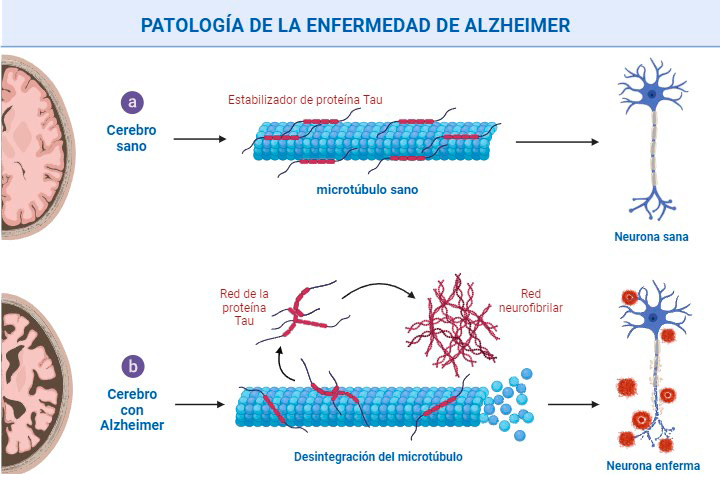
Figura 2. Patología de la enfermedad de Alzheimer
Fisiopatología
β-amiloide
El péptido β-amiloide es el principal componente de las placas seniles extracelulares características de la EA, estas se identifican en la corteza cerebral de pacientes con trastornos cognitivos y conductuales progresivos20. El péptido β-amiloide posee un peso molecular de 4 kDa y se encuentra a nivel cerebral, en personas con alteraciones cerebrales se ha encontrado en concentraciones menores que, en personas sanas21,22. La ruta que puede seguir este péptido al ser secretado se divide en dos, ambas formas se dan intracelularmente; la primera mediante el retículo endoplasmático, aparato de Golgi y endosomas; la segunda introduciéndose en la célula, gracias a la proteína que se une al receptor de la lipoproteína de baja densidad (LDL) 20.
El péptido β-amiloide es el producto principal de la proteólisis de la proteína transmembrana precursora de amiloide (PPA) cuya función exacta no está determinada, pero se supone que participa en los procesos de adhesión celular20,21,23-25. La PPA es codificada por un gen localizado en el cromosoma 21, este gen contiene 18 exones y genera 8 isoformas, las isoformas que se expresan a nivel neuronal y que contienen el exón 15 poseen una mayor capacidad de producir y liberar péptido β-amiloide. Estructuralmente la PPA posee dos dominios, estos difieren en su ubicación ya que al ser la PPA una proteína transmembrana implica un segmento extracelular denominado C-terminal y otro intracelular mucho más corto20,22,25,26.
La PPA después de haber sido sintetizada es glicosilada y empaquetada, atraviesa el citoplasma y se inserta en la membrana celular ubicada en los terminales axónicos y sinápticos22, esta proteína puede ser proteolizada y se puede dar mediante dos vías muy importantes que básicamente se diferencian en la generación o no de péptido β-amiloide y en el lugar donde se llevan a cabo. La no amiloidogénica es la vía mediante la cual no se genera péptido β-amiloide y en la que participa la enzima α-secretasa y el complejo γ-secretasa, la otra vía es la amiloidogénica, en esta sí se generan péptidos β-amiloide, se da intracelularmente después que la PPA se haya internalizado en compartimentos endosomales que contienen a la enzima β-secretasas y el complejo γ-secretasa20.22.24. El resultado de la escisión de la PPA por estas últimas enzimas genera dos especies diferentes del péptido β-amiloide, el βA40 y el βA42. Estas dos formas difieren en cuanto al lugar de la escisión y su toxicidad, el βA42 es mucho más tóxico que el βA40 y tiene la capacidad de generar con mayor facilidad las famosas placas seniles, en sí, estos dos productos van a formar oligómeros, agregados y placas20,21,25. Si este proceso se da de forma consecutiva las células encargadas de la eliminación de péptido β-amiloide no van a poder cumplir su función y esto es lo que conlleva a una acumulación de péptido β-amiloide, lo cual puede generar un proceso inflamatorio que sumado al efecto tóxico del dicho péptido lesionan las neuronas y posteriormente generan déficits sinápticos y cognitivos21,25.
Proteína Tau
La proteína Tau es la principal proteína de asociación a microtúbulos (PAM)27, es una fosfoproteína cerebral fundamental que estructuralmente está compuesta por cuatro regiones que justifican su función principal: Una región C-terminal, una región N – terminal ácida, una región rica en prolina y por último cuatro dominios repetidos (R1-4), estas dos últimas regiones se encuentran involucradas en la unión a los microtúbulos que componen el citoesqueleto de las neuronas28,29. La proteína Tau es codificada por un gen que se encuentra en el brazo largo del cromosoma 1727, en el cerebro humano este gen puede dar origen a seis diferentes isoformas de la proteína Tau, dentro de estas se tiene a la isoforma 3R Y 4R cuya proporción varía dependiendo de la patología que se padece, en el caso de la EA es de 1:127,30. La proteína Tau a nivel cerebral interactúa con los microtúbulos del axón neuronal, aproximadamente el 80% de esta proteína se une a estas estructuras en cada momento participando así de los procesos neuronales. El mecanismo mediante el cual se da esta interacción sucede en un muy corto tiempo (40 ms), lo que justifica por qué la proteína Tau no interrumpe el flujo en el proceso de transporte axonal31,32.
La proteína Tau en situaciones no patológicas se encuentra principalmente en los axones, aquí proporciona estabilidad a los microtúbulos del citoesqueleto neuronal, contribuye con el transporte de sustancia a través de las células y promueve el ensamblaje de los microtúbulos21,27, en situaciones patológicas esta proteína puede sufrir cambios, el mecanismo principal que contribuye con estas alteraciones es la fosforilación y lo hace mediante la formación de agregados neurotóxicos que se traducen en trastornos neurológicos denominados tauopatías, una de las más comunes es la EA21,28,29.
La proteína Tau puede experimentar un gran número de modificaciones postraduccionales, estas son cambios químicos que se dan después de la síntesis ribosomal, momento que es esencial ya que constituye el paso final de la síntesis proteica por tanto de la expresión génica. Estas modificaciones pueden ser: fosforilación, acetilación, glicosilación, ubiquitinación o truncamiento, siendo el primero uno de los más importantes ya que se encuentra involucrado en la patogénesis de la EA, específicamente en la generación de los famosos ovillos neurofibrilares intraneuronales característicos de esta patología21,28,30.
En la EA la proteína Tau se encuentra hiperfosforilada, esto hace que haya una agregación en forma de ovillos neurofibrilares intraneuronales debido a que una hiperfosforilación disminuye la afinidad de esta proteína por los microtúbulos, en este caso esta disociación produciría una ruptura del citoesqueleto haciendo que su estabilidad se desplome. Una consecuencia de esta disociación en experimentos in vitro según Nizynski et al. sería que la proteína Tau hiperfosforilada se una o coagregue con proteínas Tau normales29 lo cual reduciría el número de estas proteínas y su disponibilidad para cumplir su función. La agregación y coagregación conducen a una disfunción y posterior muerte neuronal, diversos hallazgos muestran que el nivel de fosforilación de la proteína Tau en personas con EA es de 3 a 4 veces mayor que personas sanas, además de haberse encontrado cerca de un 40% del total de proteínas Tau hiperfosforiladas en estos pacientes27,33.
Apolipoproteína E (ApoE)
La apolipoproteína E (ApoE) es una proteína cuyo peso molecular es de 34 kDa34, está compuesta por 299 aminoácidos y es codificada por el gen apolipoproteína E (gApoE), el cual se encuentra localizado en el brazo corto del cromosoma 1935.
La ApoE forma parte de los quilomicrones y se le considera como una proteína de unión a lípidos, estructuralmente posee dos dominios conectados mediante una unión en bisagra la cual es muy flexible; mediante el dominio N-terminal la ApoE puede unirse a los receptores y por medio del dominio C-terminal, a los lípidos21,25,33. El gApoE es el encargado de codificar esta proteína y lo hace a través de tres isoformas en seres humanos: la ApoE2, ApoE3 y ApoE4, los cuales se diferencian en distintos aspectos, ya sea estructurales, funcionales o incluso por la participación que pueden desempeñar en la patogénesis y epidemiología de la EA35,36.
La ApoE se expresa a nivel del tejido cerebral, específicamente en las células gliales con un mayor predominio de producción por parte de los astrocitos, son incluidos en las neuronas mediante las lipoproteínas de baja densidad (LDL) donde se unen con los ovillos neurofibrilares, los cuales son conglomerados proteicos característicos de los pacientes con EA25,35,37.
Fisiológicamente la ApoE cumple diversas funciones, dentro de estas se tiene que están implicadas en el transporte del colesterol necesario para el funcionamiento neuronal normal y para reparar lesiones cerebrales, lo cual hace a través de receptores ApoE ubicados en las células diana a nivel plasmático y cerebral21,33,34,39. Una vez dentro de las células diana y habiendo cumplido su función la ApoE se degrada o recicla siendo devuelta a la superficie celular. La importancia de su función radica en el uso que se le da al colesterol transportado por estas proteínas ya que este forma parte de la vaina de mielina de las neuronas, a su vez es fabricado también a nivel neuronal pero su producción disminuye con la edad de la persona debido a que, una neurona madura va a producir menos colesterol33; por tanto, será imprescindible la función que cumpla la ApoE en cuanto al mantenimiento de la actividad neuronal y con mucha más razón en personas de avanzada edad.
El transporte de colesterol a nivel cerebral depende exclusivamente de la ApoE ya que existen otros transportadores (ApoA1 y ApoB) con funciones limitadas a nivel plasmático y no a nivel cerebral34.
Las isoformas de la ApoE codificadas por el gApoE se encuentran relacionadas con la EA, específicamente una de ellas, ApoE4, está vinculada con la EA de inicio tardío. El gApoE posee tres alelos, de ellos el alelo E2 (10%) es el menos frecuente, el más común es el E3 (70% – 80%) y el relacionado con la EA, el E4 en personas sanas constituye un 20% de frecuencia en la población, pero en personas con EA se ha encontrado una frecuencia similar a la del alelo E321,33,39. La diferencia a nivel estructural de las tres isoformas es la composición de aminoácidos, cabe recalcar que la diferencia se da en una o dos posiciones de la secuencia primaria y que a pesar de ser relativamente insignificante un cambio en estas podría alterar el plegamiento estructural de la ApoE haciendo que varíe su capacidad de unión a lípidos y receptores33.
Todas las personas poseen dos alelos del gApoE, cada uno heredado por uno de los progenitores, estos pueden ser iguales (homocigotos) o diferentes (heterocigotos); de ambos grupos se ha estimado que el 91% de los individuos homocigotos, específicamente del alelo E4, desarrollarán EA en algún momento de su vida. Los no portadores del alelo E4 poseen menor riesgo de padecer EA que los portadores heterocigotos del alelo E4 (hasta 3 veces de menos riesgo) u homocigotos del alelo E4 (hasta 16 veces de menos riesgo)37,40,41.
Se ha evidenciado que las personas portadoras del alelo E4 poseen una plasticidad neuronal reducida, mielina alterada y poca capacidad de reparación tisular cerebral ante lesiones, a su vez poseen una edad de inicio más temprana de la EA41.
Fisiopatológicamente la ApoE tiene efectos sobre la degradación y eliminación del péptido β-amiloide depositado39, además sus isoformas reducen la plasticidad sináptica de manera dependiente de las mismas. En el caso del alelo E4 se ha visto involucrado en la patogénesis de la EA, debido a que regula el metabolismo, agregación y eliminación del péptido β-amiloide (βA42), principalmente se ha encontrado que el alelo E4 está relacionado con una mayor deposición y eliminación reducida del βA42, haciendo que haya una mayor agregación de este péptido, mayor neurotoxicidad, lo cual es muy característico de los individuos con EA33,39,42.
Neuroinflamación
Es una condición inflamatoria crónica del cerebro por el desequilibrio entre los mediadores proantiinflamatorios43. Además, este proceso se debe a la acumulación de microglía reactiva y astrocitos, posiblemente alojados en los depósitos de la β-amiloide21.
La microglía y los astrocitos pueden estar implicados en la neurodegeneración44,45. Las microglías son fagocitos de las placas amiloides en el SNC que se activan en respuesta a la acumulación de la β-amiloide, cambian a células ameboides, migran a las placas amiloides y liberan mediadores inflamatorios, iniciando la fagocitosis de estas46,47. Dependiendo de la etapa en la EA, cambia la función de la microglía, siendo en primera instancia neuroprotectora, luego agrava la neuroinflamación que conllevaría a una neurodegeneración44,48.
Los astrocitos son células gliales especializadas implicadas en la producción de factores neurotróficos y en el mantenimiento de la BHE. La activación prolongada conlleva a la disfunción neuronal y pérdida celular45.
Asociación entre Enfermedad periodontal y la enfermedad de Alzheimer.
Mecanismos patógenos
Antes de mencionar la patogenia de la EP, es importante recordar que la EA tiene tres etapas: preclínica, asintomática, y prodrómica, con síntomas de deterioro cognitivo leve; y demencia por EA.
La EP puede afectar cada etapa de la patogenia de la EA, pero la altera a través de mecanismos diferentes. En las primeras etapas de la EA, la EP influiría en muchos procesos como: producción y aclaramiento de la β-amiloide, fosforilación de la proteína Tau, sinapsis y funcionamiento neuronal, neurotransmisión y respuestas inmunes. En etapas avanzadas, las cargas inflamatorias como las bacterianas de la EP pueden influir más en estos procesos, pero también pueden contribuir a daños irreversibles, como la neurodegeneración y la pérdida neuronal. Entonces, la EP probablemente precede al inicio de la EA, por lo que es posible que sea causal5, 48.
Barrera hematoencefálica (BHE)
La BHE protege el cerebro, donde la mayoría de las veces se vuelve más permeable con la edad. Estas complicaciones inician en el hipocampo, un importante centro del aprendizaje y la memoria que está dañado en la EA por patógenos y factores ambientales no patógenos49,50.
En la EA existen síntomas clínicos como la pérdida de memoria y de funciones cognitivas; y eventos biológicos. Sin embargo, los eventos biológicos preceden a los síntomas clínicos, incluida la formación de la placa β-amiloide, neuroinflamación y formación de la proteína Tau21,49.
Las placas β-amiloides que se forman en el cerebro pueden inducir una respuesta inflamatoria local impulsada principalmente por células microgliales, que proliferan y liberan mediadores inflamatorios que pueden dañar las neuronas, las conexiones entre las neuronas y la BHE. Además, su salida se da a través de esta última y del líquido de las vías linfáticas. Esta respuesta inflamatoria, unido a la microbiota, que puede penetrar la BHE, pueden inducir a la fosforilación y la escisión de las proteínas Tau49.
Del mismo modo, las moléculas bacterianas inflamatorias pueden acceder al cerebro a través de diferentes rutas. Pueden ingresar al cerebro donde la BHE es permeable (órganos periventriculares), a través de: capilares fenestrados de la BHE, uso de transportadores específicos, rutas de salida de líquido cefalorraquídeo (LCR) y por aumento en la permeabilidad de la BHE5.
Mecanismos biológicos
Patógenos periodontales y productos patógenos
La EP como enfermedad inflamatoria crónica, podría tener una repercusión sistémica mediante diversos mecanismos de tipo infecciosos mediados por un grupo de microorganismos periodontales causando inflamación, dando lugar a daños cardiovasculares como cerebrales. Existe un mecanismo directo por donde la periodontitis puede conllevar a la demencia, en este mecanismo se han encontrado bacterias periodontopatógenas como Aggregatibacter actinomycetemcomitans, Porphyromonas gingivalis (inhiben la fagocitosis, expandiendo la respuesta inflamatoria crónica en la vasculatura), Tannerella forsythia, Fusobacterium nucleatum y Prevotella intermedia en tejidos cerebrales, han demostrado que tienen capacidad de invadir los tejidos cerebrales21,51.
Las especies de treponema oral como la Treponema denticola son encontradas en mayor cantidad en cerebros afectados por la EA. Treponema denticola y Porphyromonas gingivalis se encontraron de igual forma en cerebros humanos con EA pero post mortem a corto plazo, lo que podría indicar que los factores de virulencia de los patógenos mencionados tendrían un posible papel en el desarrollo de la inflamación cerebral y EA51.
Mediadores inflamatorios (IL 1, IL 6, TNFα)
En la patogenia de la EA están implicados los mecanismos inflamatorios, de forma que la inflamación y la participación de citocinas podrían ser un factor en la génesis de la EA, la inflamación es considerada un vínculo entre la periodontitis y la EA. La presencia de células gliales activadas produce un nivel significativo de proteínas inflamatorias convirtiéndose en lo más resaltante de la EA51.
La proteína Tau hiperfosforilada estimula las células gliales e inicia una neuroinflamación al producir citocinas proinflamatorias como la IL-6, TNF-α y otros factores inflamatorios como la proteína C reactiva, desencadenando una neurodegeneración. Tanto la IL – 1β y TNF-α podrían contribuir a la neuroinflamación51,52. Estudios han encontrado un aumento de citocinas proinflamatorias en pacientes ancianos con EA y periodontitis51.
Además, las células T promueven la liberación de varias citocinas y mediadores inflamatorios, donde se incluye al TNF-α e interleucinas. Estos mediadores inflamatorios, son respuesta a la infección por patógenos periodontales. Así también, las células epiteliales gingivales y los fibroblastos liberan otras citocinas y mediadores como IL-1, IL-6, TNF-α y prostaglandina E2 (PGE2)21.
Cambios vasculares
Actualmente existen estudios que detallan el gran interés de conocer la posible correlación entre enfermedad cardiovascular y periodontal, ya que el impacto de la infección oral en las enfermedades cardiovasculares no está esclarecido aún. Un epitelio subgingival inflamado y ulcerado en la periodontitis puede permitir la entrada de bacterias subgingivales y/o componentes bacterianos al torrente sanguíneo, lo que conduce al aumento y riesgo de la enfermedad cardiovascular51.
Estudios de casos y controles sobre la asociación entre la enfermedad periodontal y Alzheimer
En la tabla 1 se muestran siete estudios de casos-controles de mayor relevancia publicados sobre la asociación entre la EP y la EA. La interpretación de los resultados se encuentra limitada por la falta de consenso para definir la EP, establecer la diferencia entre demencia y la EA, clasificar los trastornos neurológicos y la selección de los pacientes. En los estudios evaluados existen multitud de definiciones de EP y de la EA, lo cual en ciertos casos podría dificultar la comparación de los resultados. El diagnóstico de EP se da mediante la evaluación clínica de ciertos parámetros periodontales utilizados en estos estudios, tales como la profundidad al sondaje (PAS), el nivel de inserción clínica (NIC), la presencia de sangrado al sondaje (SAS) el índice de placa (IP), la movilidad dental (MD) o pérdida ósea marginal (PO), estos dos últimos como métodos diagnósticos complementarios. Sin embargo, hay publicaciones que si bien utilizan alguno de estos parámetros clínicos al momento de registrarlos lo hacen en diferentes unidades, ya sea media o porcentaje.
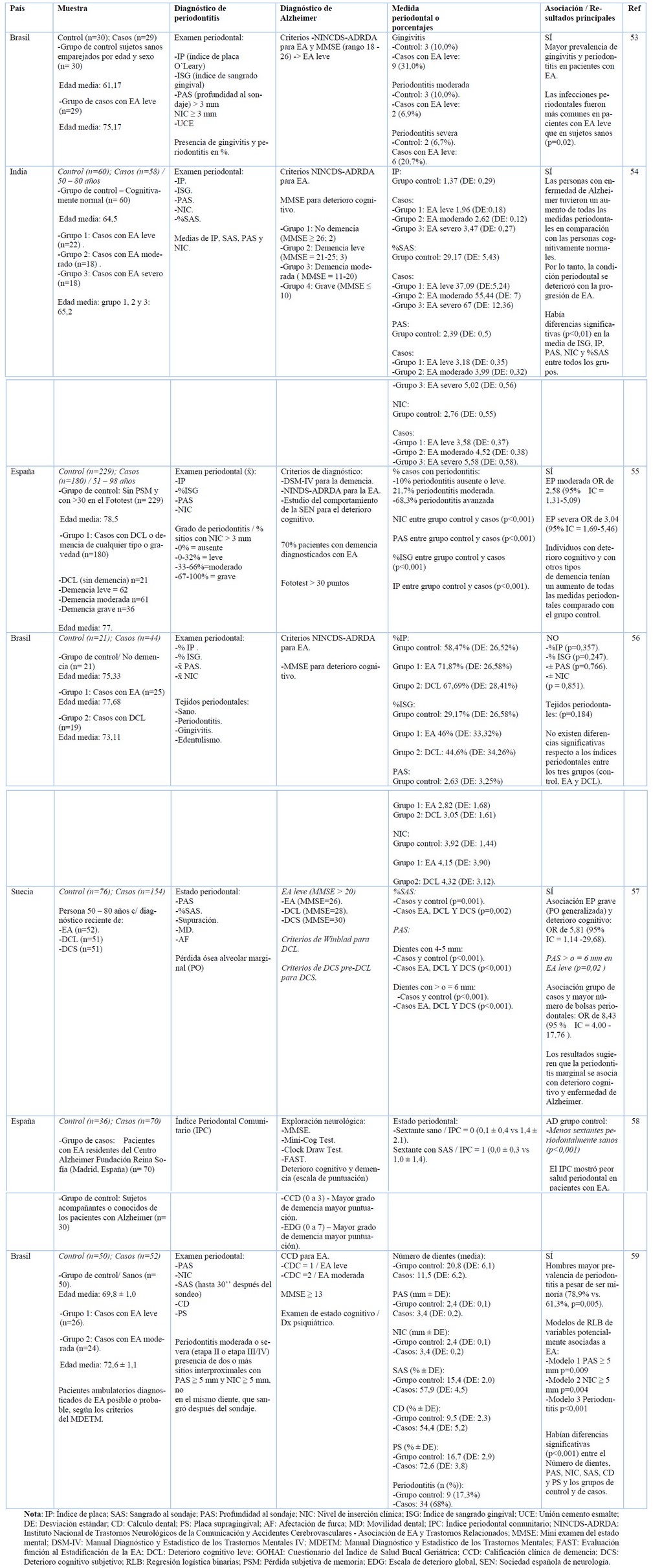
Tabla 1. Estudios de casos y controles que evalúan la asociación entre la enfermedad periodontal y la enfermedad de Alzheimer.
CONCLUSIONES
Tras una revisión de la literatura actual, los estudios demostraron que los individuos con deterioro cognitivo y con otros tipos de demencia tenían un aumento de los parámetros clínicos de la enfermedad periodontal. De esta manera se considera la posible relación entre la EP y la EA, como resultado de la alta incidencia de bacterias periodontopatógenas causantes de un estado inflamatorio crónico que podría desarrollar otras enfermedades sistémicas como: diabetes, cáncer gástrico, periodontitis, enfermedades coronarias, entre otras. Esto agrava el estado de las células del SNC disminuyendo la sinapsis interneuronal característico en una demencia temprana. Al no eliminar el factor causal de la inflamación seguirá agravándose hasta causar un estado de deterioro cognitivo mayor conocido como la EA. Se estima que hacia el 2050 más de 13 millones de personas padecerán de la EA en el mundo, esto significa que el índice de mortalidad por esta afección se elevará. Por tanto, se requieren más que avalen la relación bidireccional entre la EP y la EA, especialmente de casos y controles como de cohortes ante la imposibilidad de realizar ensayos clínicos debido a consideraciones éticas. Además, se invoca a los profesionales de salud tener en cuenta esta asociación e involucrarse cada vez más en el cuidado preventivo de la salud bucal.
Contribución de autoría: Conceptualización, A.T.G-V., S.R.V-Ch., M.A-Z., G.A.F-Q. y M.A.M-V.; metodología, M.A.M-V.; investigación, A.T.G-V., S.R.V-Ch., M.A-Z., G.A.F-Q.; recursos, A.T.G-V., S.R.V-Ch., M.A-Z., G.A.F-Q.; redacción—preparación del borrador original, A.T.G-V., S.R.V-Ch., M.A-Z., G.A.F-Q., D.R-P. y M.A.M-V.; redacción—revisión y edición, A.T.G-V., S.R.V-Ch., M.A-Z., G.A.F-Q., D.R-P. y M.A.M-V.; supervisión, M.A.M-V.; administración del proyecto, A.T.G-V. Todos los autores han leído y aceptado la versión publicada del manuscrito.
Financiamiento: Esta investigación no recibió financiación externa.
Conflicto de intereses: Los autores declaran no tener conflicto de intereses.
REFERENCIAS
1. Jellinger K, Attems J, Prevalence of dementia disorders in the oldest-old: an autopsy study. Acta Neuropathol. 2010;119:421–433.
2. Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer ‘s disease. Alzheimers Dement. Jul 2007;3(3):186-191
3. Cummings J. Alzheimer ‘s disease. N Engl J Med. 2004;351:56–67.
4. Kamer A, Craig R, Dasanayake A, Brys M, Glodzik-Sobanska L, de León M. Inflammation and Alzheimer ‘s disease: possible role of periodontal diseases. Alzheimers Dement. 2008;4(4):242-250.
5. Kamer, A, Craig, R, Niederman, R, Fortea, J, De Leon, M. Periodontal disease as a possible cause for Alzheimer’s disease. Periodontol 2000. 2020 May 8; 83(1): 242–271.
6. Borsa L, Dubois M, Sacco G, Lupi L. Analysis the Link between Periodontal Diseases and Alzheimer’s Disease: A Systematic Review. Int J Environ Res Public Health. 3 Sept 2021;18(17):9312.
7. Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimers Dement. 1 May 2018;14(3):367-429.
8. Desta N. Pathophysiological association between periodontal disease and Alzheimer’s disease: Importance of periodontal health in the elderly. J Oral Biosci. 1 Dec 2021;63(4):351-9.
9. Sureda A, Daglia M, Argüelles Castilla S, Sanadgol N, Fazel Nabavi S, Khan H, et al. Oral microbiota and Alzheimer ‘s disease: Do all roads lead to Rome? Pharmacol Res. 1 de enero de 2020;151:104582.
10. Weiss EI, Shenitzki B, Leibusor R. Coagregación microbiana en la cavidad oral. En: Kahane I., Ofek I. (eds) Towards Anti-Adhesion Therapy for Microbial Diseases. Avances en Medicina Experimental y Biología, vol 408. Springer, Boston, MA. (1996)
11. Socransky S, Haffajee A. Periodontal microbial ecology. Periodontol 2000. 2005; 38:135-87.
12. Saha S, Tomaro-Duchesneau C, Rodes L, Malhotra M, Tabrizian M, Prakash S. Investigation of probiotic bacteria as dental caries and periodontal disease biotherapeutics. Benef Microbes. 2014 Dec; 5(4):447-60.
13. Sureda A. Oral microbiota and Alzheimer’s disease: Do all roads lead to Rome? Pharmacol Res. 2020;151:104582.
14. Organización Mundial de la Salud (OMS) [Internet]. Demencia. 21 Sept 2020 [citado 8 de enero de 2022].
15. Ayodele T, Rogaeva E, Kurup J, Beecham G, Reitz C. Early-Onset Alzheimer’s Disease: What Is Missing in Research? Curr Neurol Neurosci Rep. 2021;21(2):4.
16. BrightFocus Foundation [Internet]. Enfermedad de Alzheimer: Datos y Cifras. 7 Mar 2017 [citado 8 de enero de 2022].
17. Alzheimer ‘s Association. 2021 Alzheimer ‘s disease facts and figures. Alzheimer ‘s & Dementia [Internet]. 23 Mar 2021 [citado 8 de enero del 2022]; 17(3):327-406.
18. Bondi M, Edmonds E, Salmon D. Alzheimer’s Disease: Past, Present, and Future. J Int Neuropsychol Soc. Oct 2017;23(9-10):818-31.
19. Yamazaki Y, Zhao N, Caulfield T, Liu C, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. Sept 2019;15(9):501-18.
20. Rodríguez A, Signoret V. Papel de la agregación del péptido Beta amiloide en la enfermedad de Alzheimer. Rev Educ Bioquímica [Internet]. 1 Oct 2017 [citado 25 de enero de 2022]; 36(1):2-11.
21. Liccardo D, Marzano F, Carraturo F, Guida M, Femminella G, Bencivenga L, et al. Potential Bidirectional Relationship Between Periodontitis and Alzheimer ‘s Disease. Front Physiol. 3 Jul 2020;11:683.
22. Barrera-Ocampo A, Lopera F. Amyloid-beta immunotherapy: the hope for Alzheimer disease? Colomb Med. Dic 2016;47(4):203-12.
23. Egan M, Kost J, Tariot P, Aisen P, Cummings J, Vellas B, et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N Engl J Med. 3 May 2018;378(18):1691-703.
24. Muñoz F. El péptido β-amiloide: mecanismos de neurotoxicidad – neuroprotección por antioxidantes y estrógenos. Rev Esp Geriatr Gerontol. Ener 2001;36(2):109-16.
25. Bu G. Apolipoprotein E and its receptors in Alzheimer ‘s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. May 2016;10(5):333-44.
26. Dominy S, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. Jan 2019;5(1):eaau3333.
27. Pîrşcoveanu D, Pirici I, Tudorică V, Bălşeanu T, Albu V, Bondari S, et al. Tau protein in neurodegenerative diseases – a review. Romanian J Morphol Embryol. 2017;58(4):1141-50.
28. Avila J, Lucas J, Perez M, Hernandez F. Role of tau protein in both physiological and pathological conditions. Physiol Rev. Apr 2017;84(2):361-84.
29. Niżyński B, Dzwolak W, Nieznanski K. Amyloidogenesis of Tau protein. Protein Sci Publ Protein Soc. Nov 2017;26(11):2126-50.
30. Congdon E, Sigurdsson E. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. Jul 2018;14(7):399-415.
31. Bakota L, Brandt R. Tau Biology and Tau-Directed Therapies for Alzheimer’s Disease. Drugs. Mar 2016;76(3):301-13.
32. Gauthier S, Feldman H, Schneider L, Wilcock G, Frisoni G, Hardlund J, et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet. 10 Dec 2016;388(10062):2873-84.
33. Kim J, Yoon H, Basak J, Kim J. Apolipoprotein E in synaptic plasticity and Alzheimer’s disease: potential cellular and molecular mechanisms. Mol Cells. Nov 2014;37(11):767-76.
34. Riedel B, Thompson P, Brinton R. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J Steroid Biochem Mol Biol. Jun 2016;160:134-47.
35. Barrera F, López E, Baldivieso N, Maple I, López M, Murillo Luis. Diagnóstico Actual de la Enfermedad de Alzheimer. Rev Med Clin. Jun 2018;2(2):17.
36. Waring J, Tang Q, Robieson W, King D, Das U, Dubow J, et al. APOE-ɛ4 Carrier Status and Donepezil Response in Patients with Alzheimer’s Disease. J Alzheimers Dis. Jul 2015 9;47(1):137-48.
37. Álvarez M, de la Fe A, Pedroso I, Padrón A, Álvarez M. Fisiopatología de la enfermedad de Alzheimer. Rev Mex Neuroci. May 2009;9(3):196-201.
38. Zhong N, Weisgraber K. Understanding the association of apolipoprotein E4 with Alzheimer disease: clues from its structure. J Biol Chem. Mar 2009;284(10):6027-31.
39. Rohn T. Proteolytic cleavage of apolipoprotein E4 as the keystone for the heightened risk associated with Alzheimer’s disease. Int J Mol Sci. 3 Jul 2013;14(7):14908-22.
40. Eich T, Tsapanou A, Stern Y. When time’s arrow doesn’t bend: APOE-ε4 influences episodic memory before old age. Neuropsychologia. Oct 2019;133:107180.
41. O’Donoghue M, Murphy S, Zamboni G, Nobre A, Mackay C. APOE genotype and cognition in healthy individuals at risk of Alzheimer’s disease: A review. Cortex. 1 Jul 2018;104:103-23.
42. Riedel B, Daianu M, Ver G, Mezher A, Salminen L, Galstyan A, et al. Uncovering Biologically Coherent Peripheral Signatures of Health and Risk for Alzheimer’s Disease in the Aging Brain. Front Aging Neurosci. 29 Nov 2018;10:390.
43. Femminella G, Thayanandan T, Calsolaro V, Komici K, Rengo G, et al. Imaging and molecular mechanisms of Alzheimer’s disease: a review. Int. J. Mol. Sci. Dec 2018; 19(12):3702.
44. Zuroff L, Daley D, Black K, Koronyo-Hamaoui M. Clearance of cerebral Aβ in Alzheimer’s disease: reassessing the role of microglia and monocytes. Cell Mol. Life Sci. 2017;74(12): 2167–2201.
45. Steardo L, Bronzuoli M, Iacomino A, Esposito G, Steardo L, Scuderi C. Does neuroinflammation turn on the flame in Alzheimer’s disease? Focus on astrocytes. Front. Neurosci. 2015;9(1):259.
46. Du L, Zhang Y, Chen Y, Zhu J, Yang Y, Zhang HL. Role of Microglia in Neurological Disorders and Their Potentials as a Therapeutic Target. Mol Neurobiol. Dec 2017; 54(10):7567-7584.
47. Wolf S, Boddeke H, Kettenmann H. Microglia in Physiology and Disease. Annu Rev Physiol. 10 Feb 2017;79(1):619-643.
48. Kinney J, Bemiller S, Murtishaw A, Leisgang A, Salazar A, Lamb B. Inflammation as a central mechanism in Alzheimer ‘s disease. Alzheimers Dement (N Y). 2018;4(1):575–590.
49. Ryder M, Xenoudi P. Alzheimer disease and the periodontal patient: New insights, connections, and therapies. Periodontol 2000. 2021 Oct;87(1):32–42.
50. Carter C. Genetic, Transcriptome, Proteomic, and Epidemiological Evidence for Blood-Brain Barrier Disruption and Polymicrobial Brain Invasion as Determinant Factors in Alzheimer’s Disease. J Alzheimers Dis Rep. 2017;1(1):125-157.
51. Bui F, Almeida-da-Silva C, Huynh B, Trinh A, Liu J, Woodward J, Asadi H, Ojcius DM. Association between periodontal pathogens and systemic disease. Biomed J. Feb 2019;42(1):27-35.
52. Hategan S, Kamer S, Craig R, Sinescu C, de Leon M, Jianu D, et al. Cognitive dysfunction in young subjects with periodontal disease. Neurol Sci. 2021 Nov;42(11):4511-4519.
53. de Souza R, Fabri G, Nitrini R, Anghinah R, Teixeira M, de Siqueira J, et al. Oral Infections and Orofacial Pain in Alzheimer ‘s Disease: A Case-Control Study. J. Alzheimer ‘s Dis. 1 Jan 2014;38(4):823-9.
54. Martande S, Pradeep A, Singh S, Kumari M, Suke D, Raju A, et al. Periodontal health condition in patients with Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 10 Sep 2014;29(6):498-502.
55. Gil J, Sanchez I, Carnero C, Fornieles F, Montes J, Vilchez R, et al. Is Periodontitis a Risk Factor for Cognitive Impairment and Dementia? A Case-Control Study. J Periodontol. 1 Jan 2015;86(2):244-53.
56. Cestari J, Fabri G, Kalil J, Nitrini R, Jacob W, de Siqueira J, et al. Oral Infections and Cytokine Levels in Patients with Alzheimer’s Disease and Mild Cognitive Impairment Compared with Controls. J Alzheimers Dis JAD. 19 Apr 2016;52(4):1479-85.
57. Holmer J, Eriksdotter M, Schultzberg M, Pussinen P, Buhlin K. Association between periodontitis and risk of Alzheimer’s disease, mild cognitive impairment and subjective cognitive decline: A case-control study. J Clin Periodontol. Nov 2018;45(11):1287-98.
58. Aragón F, Zea-Sevilla M, Montero J, Sancho P, Corral R, Tejedor C, et al. Oral health in Alzheimer ‘s disease: a multicenter case-control study. Clin Oral Investig. Dec 2018;22(9):3061-70.
59. De Oliveira R, Villoria G, Luiz R, Esteves J, Leão A, Feres E. Association between periodontitis and Alzheimer’s disease and its impact on the self-perceived oral health status: a case-control study. Clin Oral Investig. Feb 2021;25(2):555-62.
Received: 28 September 2023/ Accepted: 15 November 2023 / Published:15 December 2023
Citation. García-Vásquez AT, Vidal-Chávez S R, Anccasi-Zevallos M, Franco-Quispe G A, Ramos-Perfecto D y Mattos-Vela M A. Asociación entre enfermedad periodontal y enfermedad de Alzheimer. Revis Bionatura 2023;8 (4) 26. http://dx.doi.org/10.21931/RB/2023.08.04.26
Additional information Correspondence should be addressed to mmattosv@unmsm.edu.pe
Vol 9 No 4 2024

INDEXADA EN
INDEXADA EN
