
Vol 8 No 2 2023 – 54

Development of a multiplex polymerase chain reaction (m-PCR) for the detection and identification of virulent and avirulent forms of Vibrio parahaemolyticus

Hadaf Mahdi Kadhim 1,*, Colin B. Munn 2 and Martyn L. Gilpin 2,*
1 Department of Biology, College of Education, University of Al-Qadisiyah, Al-Diwaniyah, Iraq, hadaf.mahdi@qu.edu.iq
2 University of Plymouth, PL4 8AA, UK mgilpin@plymouth.ac.uk
* Correspondence: hadaf.mahdi@qu.edu.iq; Tel.: (0096416125232)
Available from: http://dx.doi.org/10.21931/RB/2023.08.02.54
ABSTRACT
This study developed a new multiplex polymerase chain reaction (m-PCR) for rapidly detecting clinically essential strains of V. parahaemolyticus. This enables the detection of total and potentially virulent strains. The m-PCR was developed by targeting the species-specific transcriptional regulator toxR gene, and sequences for an outer membrane protein and a hypothetical protein encoded by omp and htp, respectively. The omp and htp sequences were discovered originally by randomly amplified polymorphic DNA (RAPD)-PCR. The m-PCR was performed on V. parahaemolyticus isolates, 23 clinical and 32 environmental. The toxR gene 367 bp fragment amplification was found in all V. parahaemolyticus tested; 17 out of 23 clinical isolates (73.91%) showed amplification of the omp and htp. Four isolates showed amplification of the omp gene sequence but not the htp gene and 2 isolates exhibited amplification for htp but not for omp. Therefore, both sequences for omp and htp must be targeted by PCR to detect all potentially virulent strains. Of the other species tested, no amplification was seen. This study confirms that RAPD-PCR helps differentiate virulent and avirulent forms. This allowed the development of an m-PCR for identifying V. parahaemolyticus and detecting virulent forms.
Keywords: Multiplex PCR, RAPD-PCR, Vibrio parahaemolyticus
INTRODUCTION
Vibrio parahaemolyticus is a gram-negative halophilic bacterium naturally present in marine and estuarine environments throughout the temperate zone1. Pathogenic strains of this organism cause gastroenteritis after they are consumed in raw or partially cooked seafood2. However, not all strains are considered pathogenic. The presence of virulent and avirulent forms of this bacterium has led to many attempts to distinguish them and investigate potential virulence factors. Takahashi et al. 3 have reported that conventional culture methods using biochemical and nutritional tests do not differentiate virulent and avirulent strains of V. parahaemolyticus. Kim et al. 4 and Sujeewa et al. 5 have demonstrated that PCR is a valuable and rapid method for identifying V. parahaemolyticus and detecting putative virulence genes. Using PCR, detection of the tdh gene or trh gene, or both, are considered diagnostic markers for pathogenic isolates of V. parahaemolyticus6. However, these virulence genes are absent in some pathogenic strains; thus, some V. parahaemolyticus isolates that lack TDH and/or TRH remain pathogenic7-11. Furthermore, V. parahaemolyticus strains lacking tdh and/or trh caused outbreaks in Southern Chile12; therefore, there is a need to investigate other markers of virulence. Simple and validated molecular methods are available for the identification of the species V. parahaemolyticus, such as PCR for the genes encoding the transcriptional regulator (toxR), the thermolabile hemolysin (tlh) and the pR72H fragment13,4
The distinction between virulent and avirulent strains of a species may require the use of molecular typing methods; for example, randomly amplified polymorphic DNA polymerase chain reaction (RAPD–PCR) is a technique that may be used to produce a unique banding pattern to differentiate virulent strains from avirulent strains. RAPD fingerprinting has been developed as a valuable tool for distinguishing pathogenic and non-pathogenic isolates of V. harveyi14,15. RAPD uses PCR primers under low specificity (stringency) conditions16 to amplify small genome sections. An essential factor in RAPD analysis is selecting appropriate primers and optimizing PCR. The primers are simple, short, and used without prior knowledge of the template sequence of the target DNA17. RAPD-PCR may be performed with one or two primers, Hu et al. 18 used two primers in the RAPD reaction and found that the two-primer RAPD tends to produce smaller and more numerous fragments than the standard RAPD technique using only one primer. The usefulness of the two primer RAPDs is that it allows more reactions to be performed with a limited number of primers.
Previous work reported by Kadhim et al. 7 used RAPD-PCR with random primer set P1 (5’-d [GGTGCGGGAA]-3′) and P5 (5’-d [AACGCGCAAC]-3′) to reveal a unique band (approximately 600 bp) in the majority of V. parahaemolyticus clinical isolates tested, which corresponded to an outer membrane protein gene sequence (omp). This allowed the authors to develop a PCR test that amplified a 200 bp segment of omp, detecting 91% of virulent forms of V. parahaemolyticus tested. This study aimed to develop a rapid, reliable and straightforward method for differentiating virulent clinical and mainly avirulent environmental isolates of V. parahaemolyticus. This led to the development of a multiplex PCR (m-PCR) that can be used to reliably identify all isolates of the V. parahaemolyticus species and detect potentially virulent strains simultaneously. An RAPD method was used with novel sets of arbitrary primers to identify unique RAPD bands in virulent isolates. Their sequence information was used to develop the m-PCR-based method.
MATERIALS AND METHODS
Bacterial strains
All isolated bacteria used in the present study are listed in Table 1. The cultures of Vibrio were grown routinely on Tryptone soya agar (TSA) (Oxoid, Basingstoke, UK) supplemented with 3 % (w/v) sodium chloride at 37°C. Stock cultures were stored on slopes of marine salts agar (MSA) at room temperature. Other bacteria were grown at their optimum temperature on Luria-Bertani (LB) medium (Difco).
Genomic DNA extraction
According to the manufacturer’s instructions, genomic DNA was extracted from bacteria using the DNeasy tissue kit (Qiagen, Ltd., UK). The concentration and purity of DNA were measured by absorbance at 260 and 280 nm using a NanoDrop 100 spectrophotometer (Lab-Tech, UK) and its integrity after extraction was determined using 0.8 % (w/v) agarose gels in TBE buffer. The DNA was stained with ethidium bromide (0.5 µg ml-1), and photographed under UV light (309nm) with a Gel Documentation system (Uvi-Tech, UK).
RAPD-PCR
All PCR amplifications were performed in a Primus 96 plus thermal cycler (MWG Biotech). RAPD-PCR was performed using RAPD Analysis Beads (Amersham Biosciences). Reactions containing a bead resuspended in a 25 µl volume to give 1 unit Amplitaq DNA polymerase and Stoffel fragment, 0.4 mM of each dNTP, 2.5 μg BSA and buffer [3mM MgCl2, 30mM KCl and 10mM Tris, (ph 8.3)]. A 25pmol (5μl) amount of the appropriate primer and DNA template (10 ng) was added to each reaction. The following temperature-cycling parameters were used: 1 cycle at 95ºC for 5 min, followed by 45 cycles at 95 ºC for 1 min, 36 ºC for 1 min and 72 ºC for 2 min. RAPD-PCR was repeated at least two times to test for reproducibility.
PCR
PCR primers VPHTP1 and VPHTP2 were designed using primer design software Primer3 (http://biotools.umassmed.edu/bioapps/primer3_www.cgi). DNA calculator (Sigma-Genosys) was used to test primers for secondary structure and primer-dimer formation. Primer sequences were then run through the Basic Local Alignment Search Tool (BLAST) software provided by the National Centre for Biotechnology Information (NCBI) (http://blast.ncbi.nlm.nih. gov/Blast.cgi)19. Primers were obtained from MWG Biotech (MWG, Germany). Each 50 µl PCR reaction contained 1x reaction buffer containing 1.5 mM MgCl2 (Roche Applied Science, UK), 50 pmol of each primer, each deoxynucleoside triphosphate at a concentration of 0.2 mM, 1 U of Taq DNA polymerase, 20 ng of bacterial DNA and the reaction volume was made up to 50 µl using PCR grade water. PCR conditions were as follows: one cycle consisting of 94°C for 5 min, 30 cycles consisting of 94°C for 1 min, 59°C for 1 min, and 70°C for 2 min, and a final cycle consisting of 70°C for 5 min. The PCR products were electrophoresed at 90 V for approximately 6 h using a 2 % (w/v) agarose gel. A 100-bp DNA ladder (Invitrogen, UK) as standard were used as a marker in determining the size of the amplification products. DNA bands were visualized after staining with ethidium bromide (0.5 µg ml-1) or Red gel stain and photographed under ultraviolet light (309 nm) with a Gel Documentation system (Uvi-Tech, UK., software). Band sizes were assigned by direct comparison to concurrently run DNA standards. For all tests, a PCR mixture without DNA was used as negative control and to monitor for contamination.
Multiplex PCR amplification
Multiplex PCR amplification was performed on genomic DNA from each of the isolates of V. parahaemolyticus, other Vibrio species and non-Vibrio species by the simultaneous addition of three primer pairs in a 50 µl reaction mixture consisting of 30 pmol of each of the oligonucleotide primers, 0.2 mM of each deoxynucleoside triphosphate, 1x reaction buffer containing 1.5 mM MgCl2 (Roche Applied Science, UK), 60 ng of DNA template, 2 U of Taq DNA Polymerase (Roche) and the reaction volume was made up to 50 µl using PCR grade water. The primer pairs used in this PCR were as follows:
VPOMP1 5’-GTCACGCGGCCAAACAAAGAGA-3’ and
VPOMP2 5’-ACCGCATATCACTGTTGGCTGGG-3’7
VPHTP1 5′-GACATCCAATCTGCACGCAAC-3′ and
VPHTP2 5′-CCTTTCGCTTCGAGCAATCA-3′ was designed in this work; and toxR1 5’-GTCTTCTGACGCAATCGTTG-3’ and
toxR2 5’-ATACGAGTGGTTGCTGTCATG-3’ 4.
The conditions for PCR were as follows: initial denaturation at 94°C for 3 min followed by 30 cycles of denaturation at 94°C for 1 min, primer annealing at 59°C for 1 min, and primer extension at 72°C for 2 min. Following the amplification cycles, a final extension was performed at 72°C for 5 min. The m-PCR products were verified: 10 µl of the amplified DNA was separated in a 1.5 % (w/v) agarose gel using Tris-Borate/EDTA (TBE) electrophoresis buffer as described above.
DNA ligation and transformation
The unique RAPD band (Z) was excised and extracted from the gel using an agarose gel extraction kit (Roche Applied Science, UK) and purified using the ‘High pure PCR Product Purification Kit’ (Roche Applied Science, UK) according to the manufacturer’s instruction. The purified DNA fragment was ligated into pGEM-T Easy vector (Promega, UK) using T4 DNA ligase and transformed into competent cells of E. coli JM109. The transformants with pGEM-T carrying the RAPD band Z fragment were cultured on LB agar containing ampicillin (100 µg ml-1), IPTG (0.5 mM), and X-Gal (80 µg ml-1) overnight at 37°C. Recombinant plasmids were isolated from white colonies using the miniprep plasmid DNA extraction Kit (BioRad, Hemel Hempstead, UK). The presence of expected size inserts within the plasmids of transformants was verified by restriction enzyme analysis. Sequencing of both strands of selected recombinant plasmids containing the insert of the expected size was performed by MWG. Using BLAST, the nucleotide sequence data were analyzed for homology to deposited sequences in the GenBank database. The nucleotide sequence data presented in this paper has been deposited in GenBank nucleotide sequence database under accession number BA000031.2.
Sequencing of PCR products
To confirm amplicons were of the targeted sequence, selected PCR products were sequenced (MWG).
RESULTS
RAPD-PCR
Initially, V. parahaemolyticus genomic DNA from two clinical isolates (C8 and C15) and two environmental isolates (E2 and E22) were screened with nine single primers OPK1-7, OPK12 and OPK18 in RAPD-PCR reactions. These primers failed to produce unique bands to differentiate the test clinical and environmental isolates of V. parahaemolyticus. However, when 30 different OPK primer pairs were used with the same selected isolates, only the OPK13 +15 pair produced a unique band with the clinical isolates Figure 1. This amplicon was named band Z. A unique band was found When the OPK13 +15 primer set was used on the complete collection of V. parahaemolyticus isolates. Most clinical isolates produced 300 bp, and most environmental forms did not produce this band (data not shown). Patterns generated by this RAPD-PCR were reproducible in assays on different occasions. The DNA from band Z was isolated and ligated into the pGEM-T Easy vector and transformed into E. coli JM109. Restriction enzyme analysis was used to verify the presence of expected size inserts within the transformants, and selected recombinant plasmids were sequenced. BLAST analysis and comparison with the genome of V. parahaemolyticus RIMD 2210633 DNA (accession no.BA000031.2) showed identity with a 312 bp segment from base pairs 1812453 to 1812764 on chromosome 1. The nucleotide sequence was analyzed and found from a gene encoding a hypothetical protein (htp). The 312 bp band Z sequence was identical to the htp gene from V. parahaemolyticus. At the protein level, HTP was similar to proteins from V. alginolyticus (accession no. ZP_01259705.1, 62% homology), and V. harveyi (accession no. YP_001444888.1, 39% homology).
htp-PCR
Based on the DNA sequence of htp, the specific primers VPHTP1 (5′-GACATCCAATCTGCACGCAAC-3′) and VPHTP2 (5′-CCTTTCGCTTCGAGCAATCA-3′) were developed in Figure 2. PCR was performed using this primer pair on the genomic DNA of V. parahaemolyticus isolates and other Vibrio and non-Vibrio species. The expected 300 bp product was found in 19 out of 23 (82.6%) clinical isolates of V. parahaemolyticus, including clinical NCIMB reference strain R1 and only 2 out of 32 (6.25 %) environmental isolates (Table 1). No band was seen with any other Vibrio and non-Vibrio species tested.
m-PCR
This new primer set and the primer set previously developed for omp 7 were used with the species-specific primer for toxR gene 4 to develop a multiplex PCR. This reliably identified all V. parahaemolyticus isolates tested and simultaneously distinguished between virulent and avirulent strains (Fig 3). The species-specific toxR gene 367 bp segment was amplified in all V. parahaemolyticus isolates tested. Of 23 clinical isolates, 23 (100%) showed the presence of either omp or htp presence. Of these, 17 (73.91%) showed amplification of both omp and htp target sequences, 21 (91%) showed amplification of the 200 bp fragment of the omp gene, and 19 (82.6%) showed amplification of the 300 bp fragment of the htp gene (Fig 3). This showed that most clinical V. parahaemolyticus isolates contained both genes. However, four clinical isolates showed amplification of the omp gene but not the htp gene, while two clinical isolates exhibited amplification of htp but not omp. Thus both omp and htp must be targeted in PCR tests to detect all virulent strains. Out of 32 environmental isolates, none contained both omp and htp. Only 3 out of 32 (9.37%) environmental isolates showed amplification for either omp (1 = 3.12%) or htp (2 = 6.25%) genes. No amplification of omp or htp sequences was seen in other Vibrio or non-Vibrio species tested.
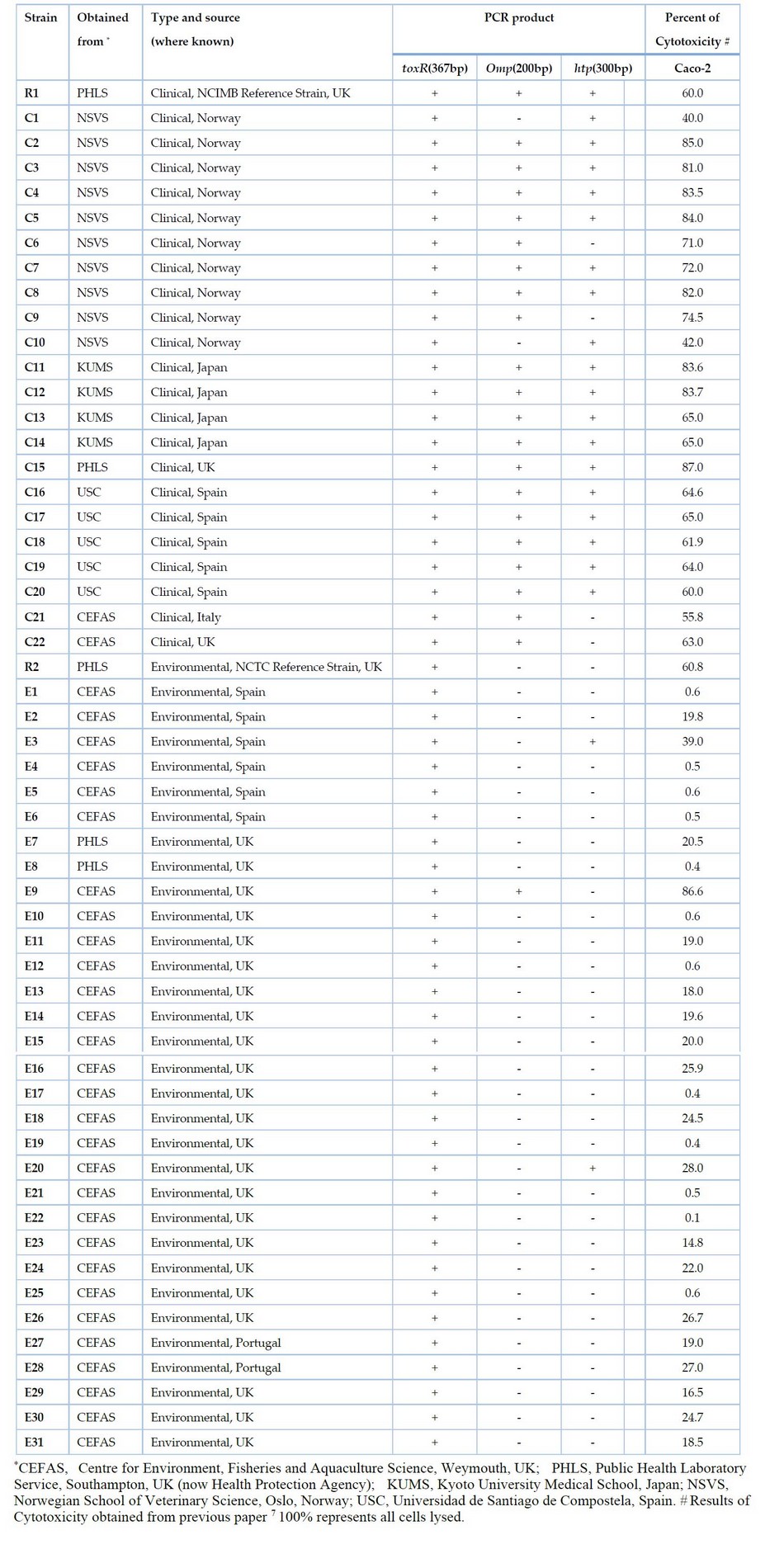
Table 1. List of Vibrio parahaemolyticus isolates tested in this study and results of multiplex PCR
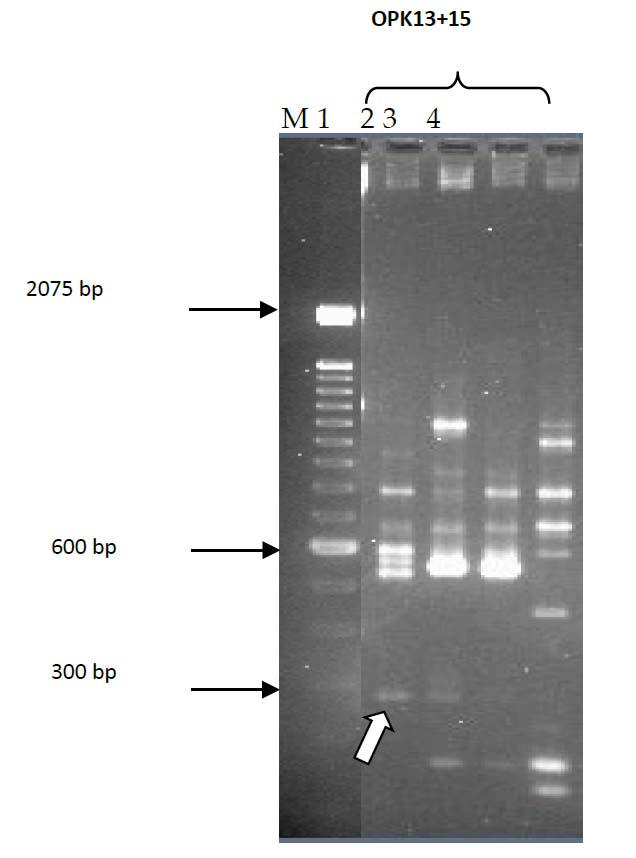
Figure 1. RAPD double primers screening of clinical and environmental isolates; Lane 1, C8; Lane 2, C15; Lane 3, E2; Lane 4, E22; Lane M, 100bp DNA ladder. Arrow indicates Band Z.

Figure 2. Nucleotide sequence of clinical V. parahaemolyticus isolates that produced band Z and locations of PCR primers VPhtp1 and VPhtp2
M 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 M
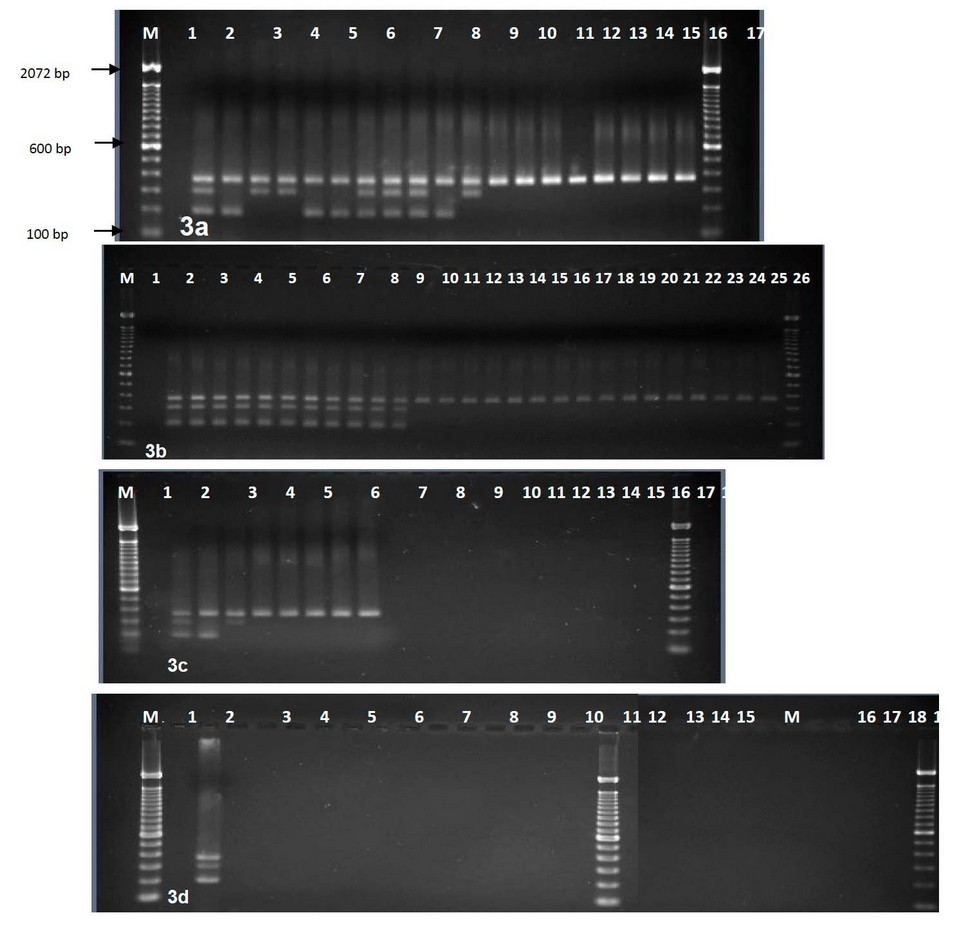
Figure 3. Agarose gel electrophoresis of multiplex PCR amplified products using three primer pairs, Lanes M, 100-bp DNA ladder. (a) Lane 1, control negative; 2-10 clinical isolates(2, R1;3, C9; 4, C1; 5, C10; 6, C21; 7, C22; 8, C7; 9, C18; 10, C2); 11-20 environmental isolates (11, E9; 12, E3; 13, E1; 14, E2; 15, E10; 16, E30; 17, E31; 18, E4; 19, E5; 20, E8). (b) Lane 1, control negative; Lanes, 2-12 clinical isolates (2, C20; 3, C19; 4, C11; 5, C12; 6, C13; 7, C14; 8, C17; 9, C16; 10, C3; 11, C4; 12, C5);Lanes, 13-28 environmental isolates (13, E11; 14, E12; 15, E16; 16, E21; 17, E22; 18, E23; 19, E24; 20, E25; 21, E26; 22, E27; 23, E28; 24, E29; 25, R2; 26, E13; 27, E14; 28, E15). (c) Lane 1, control negative; Lanes 2-3 clinical isolates (2, C8, 3, C6); Lanes 4-9 environmental isolates (4, E20; 5, E19; 6, E18; 7, E17; 8, E6; 9, E7) lanes 10-20, other Vibrio species (10, VC; 11, VM; 12-17, VV1-VV6; 18-19, VN1-VN2; 20, VS. (d) Lane 1, control negative without template DNA; 2, clinical isolate (C15); 3-13 other Vibrio species (lanes, 3-8, VA1-VA6; 9-13, VH1-VH5).; 14-17 Aeromonsa; 18-24 non Vibrio (lanes 18-19, Pf1-2; 20, St; 21, Pv; 22, Kp; 23-24, Ec1-Ec2); 25, control negative without primers.
DISCUSSION
The results of this study indicate that RAPD shows a promising ability to differentiate between clinical (virulent) and environmental (predominantly avirulent) isolates of V. parahaemolyticus. It should be noted that some environmental isolates will have the capacity to cause disease. It may be that different RAPD primer sets might be able to distinguish virulent from avirulent isolates completely and be developed into a quick PCR-based test. In the meantime, this study developed a multiplex PCR to reveal potentially virulent forms by detecting either omp (200 bp) or htp (300 bp) sequences or both; and identification of all V. parahaemolyticus isolates by producing species-specific fragment toxR gene (367 bp). The multiplex PCR method was simultaneously used to identify V. parahaemolyticus and target both omp and htp genes in a single PCR reaction. Previous work by Kadhim et al. 7 tested V. parahaemolyticus isolates for cytotoxicity to the human Caco-2 cell line and found that positive isolates for omp were strongly associated with cytotoxicity. In the current study, it should be noted that isolates carrying htp were also associated with cytotoxicity (Table 1). Isolates that did not carry either omp or htp, i.e., most of the environmental isolates, showed low cytotoxicity, except for the NCTC reference strain R2. This is environmentally sourced, did not give a positive PCR for either omp or htp, but showed a level of cytotoxicity on a par with many of the clinical (virulent ) isolates tested here. It is likely that either omp or htp, or both, act merely as markers for cytotoxicity, i.e., is not directly responsible for cytotoxicity. Perhaps they are located on a genetic region or element that carries a gene or genes for cytotoxic products when acquired. Maybe in R2, the marker genes have become detached and lost from any cytotoxic genes, but the latter remain and are active. Alternatively, R2 may contain a new or separate cytotoxic activity unrelated to omp or htp. Finally, it is possible that mutation at any of the primer-targeted regions for omp or htp sequences could have occurred, and therefore PCR amplification might fail.
Previously, between 6%20 and 9%21 of environmental V. parahaemolyticus isolates have been designated as potentially pathogenic, so the current study’s findings align with others. Further work is required to investigate whether omp or htp are virulence factors or merely virulence markers.
CONCLUSIONS
The novel multiplex PCR developed in this study can target three gene segments simultaneously and enable the identification of V. parahaemolyticus and the detection of the potentially virulent forms of this species in a single reaction. Although the reliability of this m-PCR needs further evaluation with more clinical and environmental isolates, this PCR shows excellent promise to be used as a suitable tool to identify V. parahaemolyticus and detect potentially virulent forms. Moreover, it could be used in epidemiological studies and investigations into the abundance of potentially virulent forms of V. parahaemolyticus in various environmental habitats.
Acknowledgments: We thank Craig Baker-Austin, CEFAS Laboratories, Weymouth, Dorset, UK, for invaluable assistance with the isolation, collection and maintenance of V. parahaemolyticus. This work was supported by a grant from the Ministry of Higher Education and Scientific Research (Iraq).
REFERENCES
1. Daniels, N. A., MacKinnon, L., Bishop, R. & other authors. Vibrio parahaemolyticus infections in the United States, 1973-1998. J Infect Dis, 2000; 181, 1661 – 1666.
2. Su, Y. C. & Liu, C. Vibrio parahaemolyticus: A concern of seafood safety. Food Microbiology, 2007; 24, 549-558.
3. Takahashi, H., Iwade, Y., Konuma, H. & Hara-Kudo, Y. Development of a quantitative real-time PCR method for estimation of the total number of Vibrio parahaemolyticus in contaminated shellfish and seawater. J Food Protect , 2005; 68, 1083–1088.
4. Kim, Y. B., Okuda, J., Matsumoto, C., Takahashi, N., Hashimoto, S. & Nishibuchi, M. Identification of Vibrio parahaemolyticus strains at the species level by PCR targeted to the toxR gene. J Clin Microbiol, 1999; 37, 1173-1177.
5. Sujeewa, A. K. W., Norrakiah, A. S. & Laina, M. Prevalence of toxic genes of Vibrio parahaemolyticus in shrimps (Penaeus monodon) and culture environment. International Food Research Journal, 2009; 16, 89-95.
6. Nordstrom, J. L., Vickery, M. C. L., Blackstone, G. M., Murray, S. L. & DePaola, A. Development of a multiplex real-time PCR assay with an internal amplification control for the detection of total and pathogenic Vibrio parahaemolyticus bacteria in oysters. Appl Environ Microbiol, 2007; 73, 5840-5847.
7. Kadhim, H. M., Miah, A., Munn, C. B. & Gilpin, M. L. Development of a polymerase chain reaction (PCR) test for the detection of virulent forms of Vibrio parahaemolyticus. Eur J Clin Microbiol Infect Dis, 2011; 31, 431-439.
8. Kadhim, H. M., Miah, A., Munn, C. B. & Gilpin, M. L. Survey of clinical and environmental Vibrio parahaemolyticus isolates for putative virulence factors and cytotoxicity of extracellular products to a CHO-K1 cell line. IOP Conf. Series: Earth and Environmental Science, 2021; 790: 012006
9. Nishibuchi, M., Fasano, A., Russell, R. G. & Kaper, J. B. Enterotoxigenicity of Vibrio parahaemolyticus with and without genes encoding thermostable direct hemolysin. Infect Immun, 1992; 60, 3539-3545.
10. Park, K. S., Iida, T., Yamaichi, Y., Oyagi, T., Yamamoto, K. & Honda, T. Genetic characterization of DNA region containing the trh and ure genes of Vibrio parahaemolyticus. Infect Immun, 2000; 68, 5742-5748.
11. Hussien, S. .; Doosh, K. S. . Production And Characterization Of Β-Galactosidase Enzyme In The Plant Extract From (Ziziphus Spina-Christi) And Its Application In Milk. Journal of Life Science and Applied Research. 2021, 2, 1-8..
12. García, K., Torres, R., Uribe, P., Herna’ndez, C., Rioseco, M. L., Romero, J. & Espejo, R. T. Dynamics of clinical and environmental Vibrio parahaemolyticus strains during seafood-related summer diarrhea outbreaks in Southern Chile. Applied and Environmental Microbiology, 2009; 75, 7482–7487.
13. Bej, A. K., Patterson, D. P., Brasher, C. W., Vickery, M. C. L., Jones, D. D. & Kaysner, C. A. Detection of total and hemolysin-producing Vibrio parahaemolyticus in shellfish using multiplex PCR amplification of tl, tdh and trh. Journal of Microbiological Methods, 1999; 36, 215-225.
14. Alavandi, S. V., Manoranjita, V., Vijayan, K. K., Kalaimani, N. & Santiago, T. C. Phenotypic and molecular typing of Vibrio harveyi isolates and their pathogenicity to tiger shrimp larvae. Letters in Applied Microbiology, 2006; 43, 566-570.
15. Al-Tamimi, H. .; Al-Qaraghouli, A. A. .; Khudair, Z. .; Jbara, A. A. . Effect Of Different Concentrations Of Nano-Magnesium And Nano-Titanium Oxide On The Cumulative Mortality Of Larvae Of Two Species Of Cucurbit Fruit Fly Affiliated With The Dacus In Iraq. JLSAR 2021, 2, 9-13..
16. Welsh, J. & McClelland, M. Fingerprinting genomes using PCR with arbitrary primers. Nucl Acids Res, 1990; 18, 7213-7218.
17. Blixt, Y., Knutsson, R., Borch, E. & Rådström, P. Interlaboratory random amplified polymorphic DNA typing of Yersinia enterocolitica and Y. enterocolitica-like bacteria. International Journal of Food Microbiology, 2003; 83, 15-26.
18. Hu, J., van Eysden, J. & Quiros, C. F. Generation of DNA-based markers in specific genome regions by two-primer RAPD reactions. PCR Methods and Applications, 1995; 4, 346-351.
19. Al-Enzy, A. F. M., Saed, Z. J. M., Naser, A. S., Mohammed, Th. T., Abdulateef, S. M., Al- Khalani, F. M. H. & Abdulateef, F. M. The role of adding sodium chloride in broiler chicks diets to improve production performance and antioxidant status during heat stress. Annals of Tropical Medicine and Public Health, 2020; 23(16): 231- 612. Doi: 10.36295/ASRO.2020.231612
20. Cook, D. W., Bowers, J. C. & DePaola, A. Density of total and pathogenic (tdh+) Vibrio parahaemolyticus in Atlantic and Gulf coast molluscan shellfish at harvest J Food Protection, 2002; 65, 1873-1880.
21. Cabrera-Garcia, M. E., Vazquez-Salinas, C. & Quinones-Ramirez, E. I. Serologic and molecular characterization of Vibrio parahaemolyticus strains isolated from seawater and fish products of the Gulf of Mexico. Appl Environ Microbiol, 2004; 70, 6401 – 6406.
Received: May 15, 2023/ Accepted: June 10, 2023 / Published: June 15, 2023
Citation: Kadhim, H.M.; Munn, C.B.; Gilpin, M.L. Development of a multiplex polymerase chain reaction (m-PCR) for detection and identification of virulent and avirulent forms of Vibrio parahaemolyticus. Revis Bionatura 2023;8 (2) 54. http://dx.doi.org/10.21931/RB/2023.08.02.54
Vol 9 No 4 2024

INDEXADA EN
INDEXADA EN
